

-
 Alergia
Alergia -
 Anestesiología
Anestesiología -
 Bioquímica
Bioquímica
-
 Cardiología
Cardiología
-
 Cirugía
Cirugía
-
 Dermatología
Dermatología
-
 Endocrinología y Metabolismo
Endocrinología y Metabolismo
-
 Enfermería
Enfermería
-
 Gastroenterología
Gastroenterología
-
 Hematología
Hematología
-
 Infectología
Infectología
-
 Inmunología
Inmunología
-
 Medicina Interna
Medicina Interna
-
 Nefrología
Nefrología
-
 Neumonología
Neumonología
-
 Neurología
Neurología
-
 Nutrición
Nutrición
-
 Obstetricia y Ginecología
Obstetricia y Ginecología
-
 Odontología
Odontología
-
 Oncología
Oncología
-
 Otorrinolaringología
Otorrinolaringología
-
 Pediatría
Pediatría
-
 Salud Mental
Salud Mental
-
 Salud Pública
Salud Pública
-
 Urología
Urología
-
 Más Especialidades
Más Especialidades
- Índice
-
Acceso por especialidad
-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
-
COVID-19
-
INFORMES CIENTÍFICOS
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
-
NOTICIAS/OPINIONES
-
Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
-
Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
-
Red Científica Iberoamericana (RedCIbe)
-
Conceptos Categoricos
-
Acceso por especialidad

-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
- Covid-19
-
Informes Científicos
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
- Noticias/Opiniones
- Noticias (castellano/portugués)
- Noticias (otros idiomas)
- Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
- Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
- Red Científica Iberoamericana
- Por materia
- Por fecha
-
Conceptos Categóricos

(especial para SIIC © Derechos reservados)
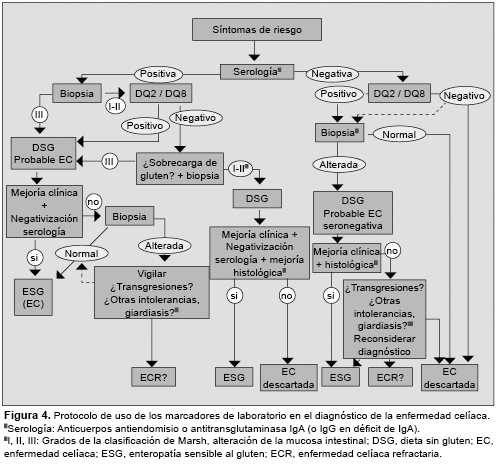
La disponibilidad de marcadores serológicos e inmunogenéticos como herramientas diagnósticas ha propiciado el avance en el conocimiento de la enfermedad celíaca y la revisión de los criterios diagnósticos.

Eduardo Arranz
Columnista Experto de SIIC
Institución:
IBGM-Universidad de Valladolid Artículos publicados por Eduardo Arranz
Eduardo Arranz*
IBGM-Universidad de Valladolid, Valladolid, España*
|
Recepción del artículo 30 de Junio, 2014 |
Aprobación 31 de Julio, 2014 |
|
Primera edición 30 de Septiembre, 2014 |
Segunda edición, ampliada y corregida 7 de Junio, 2021 |
|
|
|
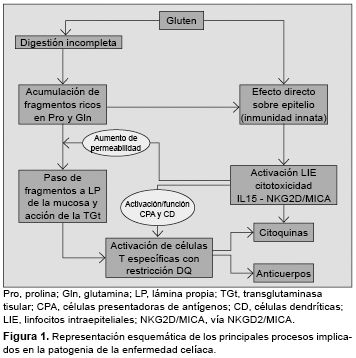
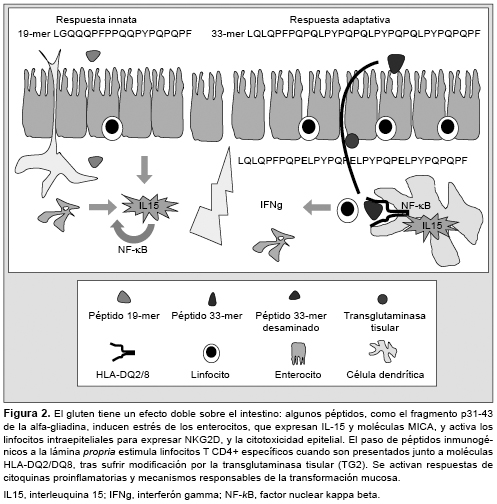
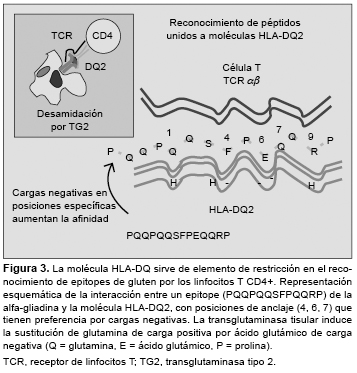
La enfermedad celíaca (EC) es un trastorno inflamatorio crónico del intestino delgado inducido por la ingestión de gluten de trigo y otras prolaminas de cereales como cebada, centeno o avena. Afecta a las personas con susceptibilidad genética, y se manifiesta por una lesión de la mucosa intestinal (con linfocitosis intraepitelial, pérdida de vellosidades y remodelación tisular), y la presencia de anticuerpos antitransglutaminasa. El modelo patogénico más aceptado se basa en la activación de una respuesta de la inmunidad adaptativa tras la estimulación de linfocitos T CD4+ mediante péptidos de gluten modificados por la enzima transglutaminasa tisular presentados junto a moléculas HLA-DQ2 o DQ8, y la producción de citoquinas y otros mediadores proinflamatorios. El gluten activa también la inmunidad innata local y mecanismos de citotoxicidad sobre el epitelio mediados por linfocitos intraepiteliales. Aunque no se conoce bien cuál es el efecto o la implicación patogénica de los anticuerpos específicos de la EC, la disponibilidad de marcadores serológicos e inmunogenéticos como herramientas diagnósticas ha propiciado el avance en el conocimiento de la EC, y la revisión de los criterios diagnósticos, especialmente en los individuos adultos con expresión mínima o atípica de la enfermedad. enfermedad celíaca, linfocitos T, péptidos de gluten, HLA-DQ2/DQ8, transglutaminasa, citoquinas, anticuerpos
Artículo completo
ASPECTOS INMUNOLOGICOS DE LA ENFERMEDAD CELIACA
1. Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol 2(9):647-55, 2002. 2. Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol 9(12):858-70, 2009. 3. Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol 29:493-525, 2011. 4. Hue S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 21(3):367-77, 2004. 5. Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 362(9377):30-7, 2003. 6. Meresse B, Curran SA, Ciszewski C, Orbelyan G, Setty M, Bhagat G, et al. Reprogramming of CTLs into natural killer-like cells in celiac disease. J Exp Med 203(5):1343-55, 2006. 7. Jabri B, Sollid LM. Mechanisms of disease: immunopathogenesis of celiac disease. Nat Clin Pract Gastroenterol Hepatol 3(9):516-25, 2006. 8. Meresse B, Ripoche J, Heyman M, Cerf-Bensussan N. Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol 2(1):8-23, 2009. 9. Tjon JM, Van Bergen J, Koning F. Celiac disease: how complicated can it get? Immunogenetics 62(10):641-51, 2010. 10. Matysiak-Budnik T, Candalh C, Dugave C, Namane A, Cellier C, Cerf-Bensussan N, et al. Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology 125(3):696-707, 2003. 11. Zimmer KP, Fischer I, Mothes T, Weissen-Plenz G, Schmitz M, Wieser H, et al. Endocytotic segregation of gliadin peptide 31-49 in enterocytes. Gut 59(3):300-10, 2010. 12. Luciani A, Villella VR, Vasaturo A, Giardino I, Pettoello-Mantovani M, Guido S, et al. Lysosomal accumulation of gliadin p31-43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut 59(3):311-9, 2010. 13. Abadie V, Discepolo V, Jabri B. Intraepithelial lymphocytes in celiac disease immunopathology. Semin Immunopathol 34(4):551-66, 2011. 14. Kagnoff MF. Celiac disease: pathogenesis of a model immunogenetic disease. J Clin Invest 117(1):41-9, 2007. 15. Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol 2(9):647-55, 2002. 16. Shewry PR, Halford NG. Cereal seed storage proteins: structures, properties and role inthe grain utilization. J Exp Bot 53:947-58, 2003. 17. Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics 64(6):455-60, 2011. 18. Arentz-Hansen H, McAdam SN, Molberg O, Fleckenstein B, Lundin KE, Jorgensen TJ, et al. Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology 123(3):803-9, 2002. 19. Vader W, Kooy Y, Van Veelen P, De Ru A, Harris D, Benckhuijsen W, et al. The gluten response in children with celiac disease is directed toward multiple gliadin and glutenin peptides. Gastroenterology 122(7):1729-37, 2002. 20. Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol 3(9):843-51, 2005. 21. Sollid LM, Thorsby E. HLA susceptibility genes in celiac disease: genetic mapping and role in pathogenesis. Gastroenterology 105(3):910-22, 1993. 22. Van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet 39(7):827-9, 2007. 23. Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med 359(26):2767-77, 2008. 24. Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural basis for gluten intolerance in celiac sprue. Science 297(5590):2275-9, 2002. 25. Menard S, Lebreton C, Schumann M, Matysiak-Budnik T, Dugave C, Bouhnik Y, et al. Paracellular versus transcellular intestinal permeability to gliadin peptides in active celiac disease. Am J Pathol 180(2):608-15, 2012. 26. Matysiak-Budnik T, Candalh C, Dugave C, Namane A, Cellier C, Cerf-Bensussan N, et al. Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology 125(3):696-707, 2003. 27. Clemente MG, De Virgiliis S, Kang JS, Macatagney R, Musu MP, Di Pierro MR, et al. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut 52(2):218-23, 2003. 28. Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 135(1):194-204 e3, 2008. 29. Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol 41(4):408-19, 2006. 30. Raki M, Tollefsen S, Molberg O, Lundin KE, Sollid LM, Jahnsen FL. A unique dendritic cell subset accumulates in the celiac lesion and efficiently activates gluten-reactive T cells. Gastroenterology 131(2):428-38, 2006. 31. Maiuri L, Ciacci C, Auricchio S, Brown V, Quaratino S, Londei M. Interleukin 15 mediates epithelial changes in celiac disease. Gastroenterology 119(4):996-1006, 2000. 32. Mention JJ, Ben Ahmed M, Begue B, Barbe U, Verkarre V, Asnafi V, et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology 125(3):730-45, 2003. 33. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21(3):357-66, 2004. 34. Maiuri L, Ciacci C, Vacca L, Ricciardelli I, Auricchio S, Quaratino S, et al. IL-15 drives the specific migration of CD94 and TCR-gammadelta intraepithelial lymphocytes in organ cultures of treated celiac patients. Am J Gastroenterol 96(1):150-6, 2001. 35. Di Sabatino A, Ciccocioppo R, Cupelli F, Cinque B, Millimaggi D, Clarkson MM, et al. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut 55(4):469-77, 2006. 36. Ebert EC. IL-15 converts human intestinal intraepithelial lymphocytes to CD94 producers of IFN-gamma and IL-10, the latter promoting Fas ligand-mediated cytotoxicity. Immunology 115(1):118-26, 2005. 37. Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 362(9377):30-7, 2003. 38. De Stefano D, Maiuri MC, Iovine B, Ialenti A, Bevilacqua MA, Carnuccio R. The role of NF-kappaB, IRF-1, and STAT-1alpha transcription factors in the iNOS gene induction by gliadin and IFN-gamma in RAW 264.7 macrophages. J Mol Med (Berl) 84(1):65-74, 2006. 39. Luciani A, Villella VR, Vasaturo A, Giardino I, Pettoello-Mantovani M, Guido S, et al. Lysosomal accumulation of gliadin p31-43 peptide induces oxidative stress and Tissue Transglutaminase mediated PPAR downregulation in intestinal epithelial cells and coeliac mucosa. Gut 59(7):1007, 2010. 40. Lundin KE, Scott H, Hansen T, Paulsen G, Halstensen TS, Fausa O, et al. Gliadin-specific, HLA-DQ(alpha 1*0501,beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med 178(1):187-96, 1993. 41. Van de Wal Y, Kooy Y, Van Veelen P, Pena S, Mearin L, Papadopoulos G, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol 161(4):1585-8, 1998. 42. Fleckenstein B, Qiao SW, Larsen MR, Jung G, Roepstorff P, Sollid LM. Molecular characterization of covalent complexes between tissue transglutaminase and gliadin peptides. J Biol Chem 279(17):17607-16, 2004. 43. Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med 4(6):713-7, 1998. 44. Arentz-Hansen H, Korner R, Molberg O, Quarsten H, Vader W, Kooy YM, et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med 191(4):603-12, 2000. 45. Hausch F, Shan L, Santiago NA, Gray GM, Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol 283(4):G996-G1003, 2002. 46. Piper JL, Gray GM, Khosla C. Effect of prolyl endopeptidase on digestive-resistant gliadin peptides in vivo. J Pharmacol Exp Ther 311(1):213-9, 2004. 47. Farstad IN, Halstensen TS, Kvale D, Fausa O, Brandtzaeg P. Topographic distribution of homing receptors on B and T cells in human gut-associated lymphoid tissue: relation of L-selectin and integrin alpha 4 beta 7 to naive and memory phenotypes. Am J Pathol 150(1):187-99, 1997. 48. Dieterich W, Storch WB, Schuppan D. Serum antibodies in celiac disease. Clin Lab 46(7-8):361-4, 2000. 49. Farstad IN, Carlsen H, Morton HC, Brandtzaeg P. Immunoglobulin A cell distribution in the human small intestine: phenotypic and functional characteristics. Immunology 101(3):354-63, 2000. 50. Qiao SW, Iversen R, Raki M, Sollid LM. The adaptive immune response in celiac disease. Semin Immunopathol 34(4):523-40, 2012. 51. Forsberg G, Hernell O, Melgar S, Israelsson A, Hammarstrom S, Hammarstrom ML. Paradoxical coexpression of proinflammatory and down-regulatory cytokines in intestinal T cells in childhood celiac disease. Gastroenterology 123(3):667-78, 2002. 52. Leon AJ, Garrote JA, Blanco-Quiros A, Calvo C, Fernandez-Salazar L, Del Villar A, et al. Interleukin 18 maintains a long-standing inflammation in coeliac disease patients. Clin Exp Immunol 146(3):479-85, 2006. 53. Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut 37(6):766-76, 1995. 54. Salvati VM, Bajaj-Elliott M, Poulsom R, Mazzarella G, Lundin KE, Nilsen EM, et al. Keratinocyte growth factor and coeliac disease. Gut 49(2):176-81, 2001. 55. Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science 307(5717):1920-5, 2005. 56. Pender SL, MacDonald TT. Matrix metalloproteinases and the gut - new roles for old enzymes. Curr Opin Pharmacol 4(6):546-50, 2004. 57. Ciccocioppo R, Di Sabatino A, Bauer M, Della Riccia DN, Bizzini F, Biagi F, et al. Matrix metalloproteinase pattern in celiac duodenal mucosa. Lab Invest 85(3):397-407, 2005. 58. Di Sabatino A, Pickard KM, Gordon JN, Salvati V, Mazzarella G, Beattie RM, et al. Evidence for the role of interferon-alfa production by dendritic cells in the Th1 response in celiac disease. Gastroenterology 133(4):1175-87, 2007. 59. Monteleone G, Pender SL, Alstead E, Hauer AC, Lionetti P, McKenzie C, et al. Role of interferon alpha in promoting T helper cell type 1 responses in the small intestine in coeliac disease. Gut 48(3):425-9, 2001. 60. Fina D, Sarra M, Caruso R, Del Vecchio Blanco G, Pallone F, Macdonald TT, et al. Interleukin-21 contributes to the mucosal t helper cell type 1 response in celiac disease. Gut 57(7):887-92, 2008. 61. Garrote JA, Gomez-Gonzalez E, Bernardo D, Arranz E, Chirdo F. Celiac disease pathogenesis: the proinflammatory cytokine network. J Pediatr Gastroenterol Nutr 47(Suppl 1):S27-32, 2008. 62. Benahmed M, Meresse B, Arnulf B, Barbe U, Mention JJ, Verkarre V, et al. Inhibition of TGF-beta signaling by IL-15: a new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology 132(3):994-1008, 2007. 63. Munz C, Steinman RM, Fujii S. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J Exp Med 202(2):203-7, 2005. 64. Calder VL, Bondeson J, Brennan FM, Foxwell BM, Feldmann M. Antigen-specific T-cell downregulation by human dendritic cells following blockade of NF-kappaB. Scand J Immunol 57(3):261-70, 2003. 65. Yu KO, Porcelli SA. The diverse functions of CD1d-restricted NKT cells and their potential for immunotherapy. Immunol Lett 100(1):42-55, 2005. 66. Van der Vliet HJ, Molling JW, Von Blomberg BM, Nishi N, Kolgen W, Van den Eertwegh AJ, et al. The immunoregulatory role of CD1d-restricted natural killer T cells in disease. Clin Immunol 112(1):8-23, 2004. 67. Stepniak D, Koning F. Celiac disease--sandwiched between innate and adaptive immunity. Hum Immunol 67(6):460-8, 2006. 68. Ribes-Koninckx C, Mearin ML, Korponay-Szabo IR, Shamir R, Husby S, Ventura A, et al. Coeliac disease diagnosis: ESPGHAN 1990 criteria or need for a change? Results of a questionnaire. J Pediatr Gastroenterol Nutr 54(1):15-9, 2011. 69. Husby S, Koletzko S, Korponay-Szabo IR, Mearin ML, Phillips A, Shamir R, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 54(1):136-60, 2012. 70. Ferguson A, Carswell F. Precipitins to dietary proteins in serum and upper intestinal secretions of coeliac children. Br Med J 1(5792):75-7, 1972. 71. Signer E, Burgin-Wolff A, Berger R, Birbaumer A, Just M. Antibodies to gliadin as a screening test for coeliac disease. A prospective study. Helv Paediatr Acta 34(1):41-52, 1979. 72. Savilahti E, Viander M, Perkkio M, Vainio E, Kalimo K, Reunala T. IgA antigliadin antibodies: a marker of mucosal damage in childhood coeliac disease. Lancet 1(8320):320-2, 1983. 73. Rostom A, Dube C, Cranney A, Saloojee N, Sy R, Garritty C, et al. The diagnostic accuracy of serologic tests for celiac disease: a systematic review. Gastroenterology 128(4 Suppl 1):S38-46, 2005. 74. Green PH, Rostami K, Marsh MN. Diagnosis of coeliac disease. Best Pract Res Clin Gastroenterol 19(3):389-400, 2005. 75. Agardh D. Antibodies against synthetic deamidated gliadin peptides and tissue transglutaminase for the identification of childhood celiac disease. Clin Gastroenterol Hepatol 5(11):1276-81, 2007. 76. Chorzelski TP, Beutner EH, Sulej J, Tchorzewska H, Jablonska S, Kumar V, et al. IgA anti-endomysium antibody. A new immunological marker of dermatitis herpetiformis and coeliac disease. Br J Dermatol 111(4):395-402, 1984. 77. Amara W, Husebekk A. Improved method for serological testing in celiac disease--IgA anti-endomysium antibody test: a comparison between monkey oesophagus and human umbilical cord as substrate in indirect immunofluorescence test. Scand J Clin Lab Invest 58(7):547-54, 1998. 78. Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med 3(7):797-801, 1997. 79. Carroccio A, Vitale G, Di Prima L, Chifari N, Napoli S, La Russa C, et al. Comparison of anti-transglutaminase ELISAs and an anti-endomysial antibody assay in the diagnosis of celiac disease: a prospective study. Clin Chem 48(9):1546-50, 2002. 80. Lewis NR, Scott BB. Systematic review: the use of serology to exclude or diagnose coeliac disease (a comparison of the endomysial and tissue transglutaminase antibody tests). Aliment Pharmacol Ther 24(1):47-54, 2006. 81. Rostami K, Kerckhaert JP, Tiemessen R, Meijer JW, Mulder CJ. The relationship between anti-endomysium antibodies and villous atrophy in coeliac disease using both monkey and human substrate. Eur J Gastroenterol Hepatol 11(4):439-42, 1999. 82. Abrams JA, Brar P, Diamond B, Rotterdam H, Green PH. Utility in clinical practice of immunoglobulin a anti-tissue transglutaminase antibody for the diagnosis of celiac disease. Clin Gastroenterol Hepatol 4(6):726-30, 2006. 83. Hadithi M, Von Blomberg BM, Crusius JB, Bloemena E, Kostense PJ, Meijer JW, et al. Accuracy of serologic tests and HLA-DQ typing for diagnosing celiac disease. Ann Intern Med 147(5):294-302, 2007. 84. Bazzigaluppi E, Roggero P, Parma B, Brambillasca MF, Meroni F, Mora S, et al. Antibodies to recombinant human tissue-transglutaminase in coeliac disease: diagnostic effectiveness and decline pattern after gluten-free diet. Dig Liver Dis 38(2):98-102, 2006. 85. Reeves GE, Squance ML, Duggan AE, Murugasu RR, Wilson RJ, Wong RC, et al. Diagnostic accuracy of coeliac serological tests: a prospective study. Eur J Gastroenterol Hepatol 18(5):493-501, 2006. 86. Abrams JA, Diamond B, Rotterdam H, Green PH. Seronegative celiac disease: increased prevalence with lesser degrees of villous atrophy. Dig Dis Sci 49(4):546-50, 2004. 87. Bonamico M, Sabbatella L, Di Tola M, Vetrano S, Ferri M, Nenna R, et al. Antiendomysial antibody detection in biopsy culture allows avoidance of gluten challenge in celiac children. J Pediatr Gastroenterol Nutr 40(2):165-9, 2005. 88. Alaedini A, Green PH. Autoantibodies in celiac disease. Autoimmunity 41(1):19-26, 2008. 89. Sardy M, Karpati S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med 195(6):747-57, 2002. 90. Hadjivassiliou M, Aeschlimann P, Strigun A, Sanders DS, Woodroofe N, Aeschlimann D. Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann Neurol 64(3):332-43, 2008. 91. Louka AS, Sollid LM. HLA in coeliac disease: unravelling the complex genetics of a complex disorder. Tissue Antigens 61(2):105-17, 2003. 92. Arranz E, Garrote JA. HLA en la enfermedad celiaca. An Pediatr Contin 2(3):163-6, 2004. 93. Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, et al. Hla types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol 64(4):469-77, 2003. 94. Polvi A, Arranz E, Fernandez-Arquero M, Collin P, Maki M, Sanz A, et al. HLA-DQ2-negative celiac disease in Finland and Spain. Hum Immunol 59(3):169-75, 1998. 95. Arranz E, Telleria JJ, Sanz A, Martin JF, Alonso M, Calvo C, et al. HLA-DQA1*0501 and DQB1*02 homozygosity and disease susceptibility in Spanish coeliac patients. Exp Clin Immunogenet 14(4):286-90, 1997. 96. Bourgey M, Calcagno G, Tinto N, Gennarelli D, Margaritte-Jeannin P, Greco L, et al. HLA related genetic risk for coeliac disease. Gut 56(8):1054-9, 2007. 97. Candore G, Lio D, Colonna Romano G, Caruso C. Pathogenesis of autoimmune diseases associated with 8.1 ancestral haplotype: effect of multiple gene interactions. Autoimmun Rev 1(1-2):29-35, 2002. 98. Louka AS, Moodie SJ, Karell K, Bolognesi E, Ascher H, Greco L, et al. A collaborative European search for non-DQA1*05-DQB1*02 celiac disease loci on HLA-DR3 haplotypes: analysis of transmission from homozygous parents. Hum Immunol 64(3):350-8, 2003. 99. Garrote JA, Arranz E, Telleria JJ, Castro J, Calvo C, Blanco-Quiros A. TNF alpha and LT alpha gene polymorphisms as additional markers of celiac disease susceptibility in a DQ2-positive population. Immunogenetics 54(8):551-5, 2002. 100. Louka AS, Lie BA, Talseth B, Ascher H, Ek J, Gudjonsdottir AH, et al. Coeliac disease patients carry conserved HLA-DR3-DQ2 haplotypes revealed by association of TNF alleles. Immunogenetics 55(5):339-43, 2003. 101. Lopez-Vazquez A, Fuentes D, Rodrigo L, Gonzalez S, Moreno M, Fernandez E, et al. MHC class I region plays a role in the development of diverse clinical forms of celiac disease in a Saharawi population. Am J Gastroenterol 99(4):662-7, 2004. 102. Bilbao JR, Martin-Pagola A, Perez De Nanclares G, Calvo B, Vitoria JC, Vazquez F, et al. HLA-DRB1 and MICA in autoimmunity: common associated alleles in autoimmune disorders. Ann N Y Acad Sci 1005:314-8, 2003. 103. Ramos-Arroyo MA, Feijoo E, Sanchez-Valverde F, Aranburu E, Irisarri N, Olivera JE, et al. Heat-shock protein 70-1 and HLA class II gene polymorphisms associated with celiac disease susceptibility in Navarra (Spain). Hum Immunol 62(8):821-5, 2001. 104. Monsuur AJ, De Bakker PI, Alizadeh BZ, Zhernakova A, Bevova MR, Strengman E, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat Genet 37(12):1341-4, 2005. 105. Holopainen P, Naluai AT, Moodie S, Percopo S, Coto I, Clot F, et al. Candidate gene region 2q33 in European families with coeliac disease. Tissue Antigens 63(3):212-22, 2004. 106. Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet 40(4):395-402, 2008. 107. Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet 42(4):295-302, 2010. 108. Eiras P, Leon F, Camarero C, Lombardia M, Roldan E, Bootello A, et al. Intestinal intraepithelial lymphocytes contain a CD3- CD7 subset expressing natural killer markers and a singular pattern of adhesion molecules. Scand J Immunol 52(1):1-6, 2000. 109. Spencer J, Isaacson PG, MacDonald TT, Thomas AJ, Walker-Smith JA. Gamma/delta T cells and the diagnosis of coeliac disease. Clin Exp Immunol 85(1):109-13, 1991. 110. Arranz E, Bode J, Kingstone K, Ferguson A. Intestinal antibody pattern of coeliac disease: association with gamma/delta T cell receptor expression by intraepithelial lymphocytes, and other indices of potential coeliac disease. Gut 35(4):476-82, 1994. 111. Leon F, Eiras P, Camarero C, Roldan E, Sanchez L, R RP, et al. Advances in the diagnosis of celiac disease: anti-transglutaminase antibodies and intestinal intraepithelial lymphocytes. Gastroenterol Hepatol 25(6):416-22, 2002. 112. Savilahti E, Ormala T, Arato A, Hacsek G, Holm K, Klemola T, et al. Density of gamma/delta T cells in the jejunal epithelium of patients with coeliac disease and dermatitis herpetiformis is increased with age. Clin Exp Immunol 109(3):464-7, 1997. 113. Leon F. Flow cytometry of intestinal intraepithelial lymphocytes in celiac disease. J Immunol Methods 363(2):177-86, 2011. 114. Moron B, Bethune MT, Comino I, Manyani H, Ferragud M, Lopez MC, et al. Toward the assessment of food toxicity for celiac patients: characterization of monoclonal antibodies to a main immunogenic gluten peptide. PLoS One 3(5):e2294, 2008. 115. Comino I, Real A, Vivas S, Siglez MA, Caminero A, Nistal E, et al. Monitoring of gluten-free diet compliance in celiac patients by assessment of gliadin 33-mer equivalent epitopes in feces. Am J Clin Nutr 95(3):670-7, 2012. 116. Walker MM, Murray JA. An update in the diagnosis of coeliac disease. Histopathology 59(2):166-79, 2011. 117. Tack GJ, Verbeek WH, Schreurs MW, Mulder CJ. The spectrum of celiac disease: epidemiology, clinical aspects and treatment. Nat Rev Gastroenterol Hepatol 7(4):204-13, 2010. 118. Duerksen DR, Wilhelm-Boyles C, Parry DM. Intestinal permeability in long-term follow-up of patients with celiac disease on a gluten-free diet. Dig Dis Sci 50(4):785-90, 2005. 119. Ertekin V, Selimoglu MA, Turgut A, Bakan N. Fecal calprotectin concentration in celiac disease. J Clin Gastroenterol 44(8):544-6, 2010. 120. Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther 26(5):757-66, 2007. 121. Pyle GG, Paaso B, Anderson BE, Allen DD, Marti T, Li Q, et al. Effect of pretreatment of food gluten with prolyl endopeptidase on gluten-induced malabsorption in celiac sprue. Clin Gastroenterol Hepatol 3(7):687-94, 2005. 122. Daveson AJ, Jones DM, Gaze S, McSorley H, Clouston A, Pascoe A, et al. Effect of hookworm infection on wheat challenge in celiac disease--a randomised double-blinded placebo controlled trial. PLoS One 6(3):e17366, 2011. 123. Crespo Perez L, Castillejo de Villasante G, Cano Ruiz A, Leon F. Non-dietary therapeutic clinical trials in coeliac disease. Eur J Intern Med 23(1):9-14, 2012. Artículos publicados por el autor (selección): Definitions and diagnostic criteria of latent and potential coeliac disease. Common Food Intolerances I: Epidemiology of Coeliac Disease. Karger 2:119-127, 1992 Clinical and pathological spectrum of coeliac disease Gut 34:150-151, 1993 Secretory antibody pattern of coeliac disease: occurrence in patients with normal jejunal biopsy histology Gastroenterology 104:1263-1272, 1993 Gamma/delta T cell receptor expression by intra-epithelial lymphocytes in relation to other indices of potential coeliac disease Gut 35:476-482, 1994 The pattern of cytokine expresión determines the degree of mucosal damage Gut 56(56):441-443, 2007 |
|

|
Está expresamente prohibida la redistribución y la redifusión de todo o parte de los contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin previo y expreso consentimiento de SIIC. |

