

-
 Alergia
Alergia -
 Anestesiología
Anestesiología -
 Bioquímica
Bioquímica
-
 Cardiología
Cardiología
-
 Cirugía
Cirugía
-
 Dermatología
Dermatología
-
 Endocrinología y Metabolismo
Endocrinología y Metabolismo
-
 Enfermería
Enfermería
-
 Gastroenterología
Gastroenterología
-
 Hematología
Hematología
-
 Infectología
Infectología
-
 Inmunología
Inmunología
-
 Medicina Interna
Medicina Interna
-
 Nefrología
Nefrología
-
 Neumonología
Neumonología
-
 Neurología
Neurología
-
 Nutrición
Nutrición
-
 Obstetricia y Ginecología
Obstetricia y Ginecología
-
 Odontología
Odontología
-
 Oncología
Oncología
-
 Otorrinolaringología
Otorrinolaringología
-
 Pediatría
Pediatría
-
 Salud Mental
Salud Mental
-
 Salud Pública
Salud Pública
-
 Urología
Urología
-
 Más Especialidades
Más Especialidades
- Índice
-
Acceso por especialidad
-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
-
COVID-19
-
INFORMES CIENTÍFICOS
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
-
NOTICIAS/OPINIONES
-
Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
-
Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
-
Red Científica Iberoamericana (RedCIbe)
-
Conceptos Categoricos
-
Acceso por especialidad

-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
- Covid-19
-
Informes Científicos
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
- Noticias/Opiniones
- Noticias (castellano/portugués)
- Noticias (otros idiomas)
- Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
- Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
- Red Científica Iberoamericana
- Por materia
- Por fecha
-
Conceptos Categóricos

Casos Clínicos
ARTROGRIPOSIS DISTAL: INFORME DE UN CASO SUGESTIVO DE SINDROME DE FREEMAN-SHELDON
Las artrogriposis distales son un grupo de malformaciones autosómicas dominantes que se presentan en uno de cada 3 000 nacidos vivos y se caracterizan por la presentación de contracturas musculares congénitas en una o dos más áreas del cuerpo.
Clasificación en siicsalud
Artículos originales > Expertos del Mundo >
página /dato/casiic.php/144940
Especialidades



Artículos originales > Expertos del Mundo >
página /dato/casiic.php/144940
Especialidades
Primera edición en siicsalud
4 de mayo, 2015
4 de mayo, 2015
ARTROGRIPOSIS DISTAL: INFORME DE UN CASO SUGESTIVO DE SINDROME DE FREEMAN-SHELDON
(especial para SIIC © Derechos reservados)
(especial para SIIC © Derechos reservados)
Introducción
El síndrome de Freeman-Sheldon con código MIM (Mendelian Inheritance in Man): 193700, es un tipo de artrogriposis distal asociado con contracturas musculares a nivel orofacial que se manifiesta con unas características facies “silbantes”. Este se hereda generalmente de forma autosómica dominante, aunque hay informes de herencia recesiva y ligada al cromosoma X, y tiene una expresividad variable. Se debe a mutaciones en el gen MYH3 que alteran la función normal inhibitoria de la troponina I. Esta entidad tiene un buen pronóstico después de superar las primeras barreras asociadas con dificultades para la alimentación, el crecimiento y el desarrollo del lenguaje. El tratamiento se basa principalmente en la corrección quirúrgica de las limitaciones, lo que a su vez representa un alto riesgo, debido a las posibles complicaciones del procedimiento anestésico.1
Se presenta el caso de una paciente de cinco meses de edad que presenta artrogriposis distal y facies que sugieren síndrome de Freeman-Sheldon. Caso clínico
Paciente de cinco meses de edad, hija de madre de 33 años, y padre de 38 años, producto de tercer embarazo, con antecedente de haber tenido dos abortos espontaneas. Informe de disminución de movimientos fetales en el último mes de embarazo, y oligohidramnios en las últimas ecografías, por lo que se programó parto por cesárea. Nació de 34 semanas con un peso de 1 280 gramos, pequeño para la edad gestacional, y una talla de 37.5 cm.
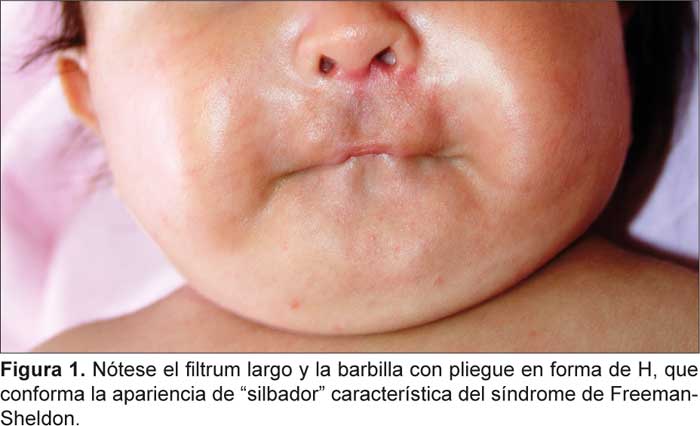
Al examen físico presenta frente estrecha, orejas displásicas con rotación posterior, telecanto, huesos nasales hipoplásicos, orificios nasales estrechos, facies silbantes, surco en H sobre el mentón y ligera microrretrognatia. A nivel de las manos, disminución de dermatoglifos, desviación ulnar de los dedos con superposición del segundo sobre el tercero y el quinto sobre el cuarto; en los pies, superposición del segundo sobre el tercer artejo del pie izquierdo y fosa sacra. En los exámenes complementarios se encontró cariotipo 46, XX (700 bandas, bandeo G) y el electroencefalograma se hallaba dentro de los límites normales. Discusión
Las artrogriposis distales son un grupo de malformaciones autosómicas dominantes que se presentan en uno de cada 3 000 nacidos vivos, se caracterizan por la presentación de contracturas musculares congénitas en dos o más áreas del cuerpo, asociadas con un rasgo constante que consiste en el compromiso de manos y pies.1 Este grupo puede ser dividido en hasta doce tipos diferentes con características variables. En 1938, Freeman y Sheldon describieron el síndrome de cara silbante, conocido ahora como síndrome de Freeman-Sheldon o como artrogriposis distal tipo 2. En ésta, el compromiso muscular se da principalmente a nivel facial, manifestándose en unas facies características que se destacan por presentar boca pequeña, pliegues nasolabiales prominentes y un hoyuelo en forma de H en el mentón. En asociación con las contracturas faciales se presentan los rasgos típicos en manos y pies, que incluyen una desviación cubital de la muñeca y dedos, camptodactilia, pliegues de flexión hipoplásicos, superposición de dedos y pie equino varo.2 En adición a estos signos, algunas características frecuentes en el síndrome de Freeman-Sheldon son escoliosis, blefarofimosis, ptosis, estrabismo y apiñamiento dental.1-3
Como parte del grupo de las artrogriposis distales, el síndrome de Freeman-Sheldon se hereda de forma autosómica dominante; presenta una penetrancia variable con informes de múltiples portadores asintomáticos, así como de casos esporádicos.3-5 Se debe a una mutación en el gen MYH3 que causa alteraciones en el segmento C-terminal de la proteína sarcomérica troponina I, se han identificado más de 26 mutaciones en este gen ubicado en el cromosoma 17p13.1 asociadas con la presentación de este síndrome.3,4 Entre las artrogriposis distales, existe otra muy similar al síndrome de Freeman-Sheldon, conocida como síndrome de Sheldon-Hall o artrogriposis distal 2B. En esta patología las manifestaciones son más leves, sin presentar el típico hoyuelo en H y menos pronunciamiento de los pliegues nasolabiales.6 En el caso de esta última se han encontrado, además de mutaciones en el gen MYH3, mutaciones en el TNNI2, aunque se desconoce el mecanismo exacto por el cual genera la contractura.4 En 2013, Li y colaboradores comunicaron la presencia de mutaciones del gen TNNI2 en pacientes con síndrome de Freeman-Sheldon clásico, lo cual sugiere que estos dos síndromes podrían representar en realidad un continuo fenotípico.5,7
El diagnóstico es inicialmente clínico, dada la presentación tan clásica del síndrome; sin embargo, los estudios complementarios, además del análisis molecular, incluyen electromiografía y biopsia de músculo, la cual revela generalmente fibrosis que contribuye al desarrollo de las contracturas.1,6 Igualmente, deben evaluarse de forma completa los compromisos asociados con la enfermedad mediante estudios imagenológicos, incluyendo radiografías óseas y valoración de espina bífida por ecografía, dada la alta incidencia de este cuadro en los pacientes con síndrome de Freeman-Sheldon, y estudios funcionales, como por ejemplo de función pulmonar, teniendo en cuenta las repercusiones asociadas con malformaciones musculo-esqueléticas.6,8 En el diagnóstico diferencial se consideran los síndromes con microstomía y los que cursan con artrogriposis, como la distrofia muscular congénita, miopatía miotubular, síndrome de Schwartz-Jampel tipo 1b, síndrome de Stuve-Wiedemann, entre otros.2 Las principales complicaciones asociadas con este síndrome se relacionan con las malformaciones faciales, que implican que desde los primeros meses se presenten dificultades para la alimentación y, por consiguiente, para el adecuado crecimiento. Además, se presentan dificultades para el desarrollo del lenguaje, las posibles complicaciones en cuanto a la higiene oral y el riesgo de broncoaspiración; la función cognitiva y el desarrollo psicomotor se encuentran generalmente conservados.2,9 El abordaje más importante en estos pacientes es la corrección quirúrgica de los defectos, incluyendo la corrección de la microstomía, los problemas oculares y nasales, y la corrección de defectos ortopédicos entre otras cirugías que puedan ser necesarias.9 Este abordaje representa un reto a su vez, en especial desde el punto de vista de los anestesiólogos debido a las dificultades para la intubación y venopunción; asimismo, como por la limitación del uso de relajantes musculares por tratarse de una miopatía.10,11 Por otra parte, la miopatía generalizada se asocia la aparición de cifoescoliosis, que en adición a la miopatía de los músculos intercostales puede llevar a enfermedad pulmonar restrictiva, por lo que debe mantenerse también un control estricto en este aspecto.8 Una vez superadas las dificultades anteriores, estos pacientes tienen un buen pronóstico, sin dejar de lado la posibilidad de recurrencia de la microstomía, lo que implica manejo por terapia, y el uso de dispositivos de estiramiento.8 En la mayoría de los casos, la corrección temprana de las deformidades resulta en una calidad de vida adecuada, con una expectativa de vida normal.12
El síndrome de Freeman-Sheldon con código MIM (Mendelian Inheritance in Man): 193700, es un tipo de artrogriposis distal asociado con contracturas musculares a nivel orofacial que se manifiesta con unas características facies “silbantes”. Este se hereda generalmente de forma autosómica dominante, aunque hay informes de herencia recesiva y ligada al cromosoma X, y tiene una expresividad variable. Se debe a mutaciones en el gen MYH3 que alteran la función normal inhibitoria de la troponina I. Esta entidad tiene un buen pronóstico después de superar las primeras barreras asociadas con dificultades para la alimentación, el crecimiento y el desarrollo del lenguaje. El tratamiento se basa principalmente en la corrección quirúrgica de las limitaciones, lo que a su vez representa un alto riesgo, debido a las posibles complicaciones del procedimiento anestésico.1
Se presenta el caso de una paciente de cinco meses de edad que presenta artrogriposis distal y facies que sugieren síndrome de Freeman-Sheldon. Caso clínico
Paciente de cinco meses de edad, hija de madre de 33 años, y padre de 38 años, producto de tercer embarazo, con antecedente de haber tenido dos abortos espontaneas. Informe de disminución de movimientos fetales en el último mes de embarazo, y oligohidramnios en las últimas ecografías, por lo que se programó parto por cesárea. Nació de 34 semanas con un peso de 1 280 gramos, pequeño para la edad gestacional, y una talla de 37.5 cm.
Al examen físico presenta frente estrecha, orejas displásicas con rotación posterior, telecanto, huesos nasales hipoplásicos, orificios nasales estrechos, facies silbantes, surco en H sobre el mentón y ligera microrretrognatia. A nivel de las manos, disminución de dermatoglifos, desviación ulnar de los dedos con superposición del segundo sobre el tercero y el quinto sobre el cuarto; en los pies, superposición del segundo sobre el tercer artejo del pie izquierdo y fosa sacra. En los exámenes complementarios se encontró cariotipo 46, XX (700 bandas, bandeo G) y el electroencefalograma se hallaba dentro de los límites normales. Discusión
Las artrogriposis distales son un grupo de malformaciones autosómicas dominantes que se presentan en uno de cada 3 000 nacidos vivos, se caracterizan por la presentación de contracturas musculares congénitas en dos o más áreas del cuerpo, asociadas con un rasgo constante que consiste en el compromiso de manos y pies.1 Este grupo puede ser dividido en hasta doce tipos diferentes con características variables. En 1938, Freeman y Sheldon describieron el síndrome de cara silbante, conocido ahora como síndrome de Freeman-Sheldon o como artrogriposis distal tipo 2. En ésta, el compromiso muscular se da principalmente a nivel facial, manifestándose en unas facies características que se destacan por presentar boca pequeña, pliegues nasolabiales prominentes y un hoyuelo en forma de H en el mentón. En asociación con las contracturas faciales se presentan los rasgos típicos en manos y pies, que incluyen una desviación cubital de la muñeca y dedos, camptodactilia, pliegues de flexión hipoplásicos, superposición de dedos y pie equino varo.2 En adición a estos signos, algunas características frecuentes en el síndrome de Freeman-Sheldon son escoliosis, blefarofimosis, ptosis, estrabismo y apiñamiento dental.1-3
Como parte del grupo de las artrogriposis distales, el síndrome de Freeman-Sheldon se hereda de forma autosómica dominante; presenta una penetrancia variable con informes de múltiples portadores asintomáticos, así como de casos esporádicos.3-5 Se debe a una mutación en el gen MYH3 que causa alteraciones en el segmento C-terminal de la proteína sarcomérica troponina I, se han identificado más de 26 mutaciones en este gen ubicado en el cromosoma 17p13.1 asociadas con la presentación de este síndrome.3,4 Entre las artrogriposis distales, existe otra muy similar al síndrome de Freeman-Sheldon, conocida como síndrome de Sheldon-Hall o artrogriposis distal 2B. En esta patología las manifestaciones son más leves, sin presentar el típico hoyuelo en H y menos pronunciamiento de los pliegues nasolabiales.6 En el caso de esta última se han encontrado, además de mutaciones en el gen MYH3, mutaciones en el TNNI2, aunque se desconoce el mecanismo exacto por el cual genera la contractura.4 En 2013, Li y colaboradores comunicaron la presencia de mutaciones del gen TNNI2 en pacientes con síndrome de Freeman-Sheldon clásico, lo cual sugiere que estos dos síndromes podrían representar en realidad un continuo fenotípico.5,7
El diagnóstico es inicialmente clínico, dada la presentación tan clásica del síndrome; sin embargo, los estudios complementarios, además del análisis molecular, incluyen electromiografía y biopsia de músculo, la cual revela generalmente fibrosis que contribuye al desarrollo de las contracturas.1,6 Igualmente, deben evaluarse de forma completa los compromisos asociados con la enfermedad mediante estudios imagenológicos, incluyendo radiografías óseas y valoración de espina bífida por ecografía, dada la alta incidencia de este cuadro en los pacientes con síndrome de Freeman-Sheldon, y estudios funcionales, como por ejemplo de función pulmonar, teniendo en cuenta las repercusiones asociadas con malformaciones musculo-esqueléticas.6,8 En el diagnóstico diferencial se consideran los síndromes con microstomía y los que cursan con artrogriposis, como la distrofia muscular congénita, miopatía miotubular, síndrome de Schwartz-Jampel tipo 1b, síndrome de Stuve-Wiedemann, entre otros.2 Las principales complicaciones asociadas con este síndrome se relacionan con las malformaciones faciales, que implican que desde los primeros meses se presenten dificultades para la alimentación y, por consiguiente, para el adecuado crecimiento. Además, se presentan dificultades para el desarrollo del lenguaje, las posibles complicaciones en cuanto a la higiene oral y el riesgo de broncoaspiración; la función cognitiva y el desarrollo psicomotor se encuentran generalmente conservados.2,9 El abordaje más importante en estos pacientes es la corrección quirúrgica de los defectos, incluyendo la corrección de la microstomía, los problemas oculares y nasales, y la corrección de defectos ortopédicos entre otras cirugías que puedan ser necesarias.9 Este abordaje representa un reto a su vez, en especial desde el punto de vista de los anestesiólogos debido a las dificultades para la intubación y venopunción; asimismo, como por la limitación del uso de relajantes musculares por tratarse de una miopatía.10,11 Por otra parte, la miopatía generalizada se asocia la aparición de cifoescoliosis, que en adición a la miopatía de los músculos intercostales puede llevar a enfermedad pulmonar restrictiva, por lo que debe mantenerse también un control estricto en este aspecto.8 Una vez superadas las dificultades anteriores, estos pacientes tienen un buen pronóstico, sin dejar de lado la posibilidad de recurrencia de la microstomía, lo que implica manejo por terapia, y el uso de dispositivos de estiramiento.8 En la mayoría de los casos, la corrección temprana de las deformidades resulta en una calidad de vida adecuada, con una expectativa de vida normal.12
María Fernanda Hernandez-Amaris, Universidad ICESI Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras (CIACER), CALI, Colombia,
e-mail: hmpachajoa@icesi.edu.co
1. Stevenson DA, Carey JC, Palumbos J, Rutherford A, Dolcourt J, Bamshad MJ. Clinical characteristics and natural history of Freeman-Sheldon syndrome. Pediatrics 117(3):754-62, 2006.
2. Ashish J, Muralidhar R, Vijayalakshmi P, Meenakshi S. Freeman-Sheldon syndrome: case report and review of the literature. Int Ophthalmol 31(5):405-7, 2011.
3. Tajsharghi H, Kimber E, Kroksmark AK, Jerre R, Tulinius M, Oldfors A. Embryonic myosin heavy-chain mutations cause distal arthrogryposis and developmental myosin myopathy that persists postnatally. Arch Neurol 65(8):1083-90, 2008.
4. Kimber E, Tajsharghi H, Kroksmark AK, Oldfors A, Tulinius M. Distal arthrogryposis: clinical and genetic findings. Acta Paediatr 101(8):877-87, 2012.
5. Shrimpton AE, Hoo JJ. A TNNI2 mutation in a family with distal arthrogryposis type 2B. Eur J Med Genet 49(2):201-6, 2006.
6. Ferrari D, Bettuzzi C, Donzelli O. Freeman-Sheldon syndrome. A case report and review of the literature. Chir Organi Mov 92(2):127-31, 2008.
7. Li X, Jiang M, Han W, Zhao N, Liu W, Sui Y, et al. A novel TNNI2 mutation causes Freeman-Sheldon syndrome in a Chinese family with an affected adult with only facial contractures. Gene 527(2):630-5, 2013.
8. Attia A, Suleman M, Nwasser AAAA. Freeman-Sheldon syndrome with respiratory failure: A case report. Respiratory Medicine CME 1(4):274-7, 2008.
9. Sadrimanesh R, Hassani A, Vahdati SA, Chaghari H, Sadr-Eshkevari P, Rashad A. Freeman-Sheldon syndrome: combined surgical and non-surgical approach. J Craniomaxillofac Surg 41(5):397-402, 2013.
10. Madi-Jebara S, El-Hajj C, Jawish D, Ayoub E, Kharrat K, Antakly MC. Anesthetic management of a patient with Freeman-Sheldon syndrome: case report. J Clin Anesth 19(6):460-2, 2007.
11. Patel K, Gursale A, Chavan D, Sawant P. Anaesthesia challenges in Freeman-Sheldon syndrome. Indian J Anaesth 57(6):632-3, 2013.
12. Malkawi H, Tarawneh M. The whistling face syndrome, or craniocarpotarsal dysplasia. Report of two cases in a father and son and review of the literature. J Pediatr Orthop 3(3):364-9, 1983.
Está expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC.
ua91218

