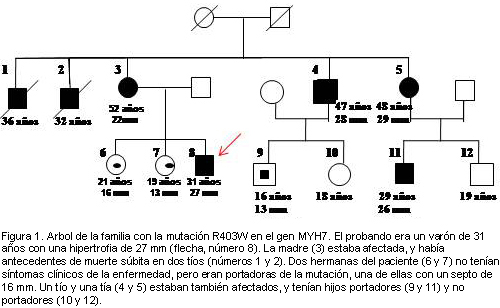
Bibliografía del artículo
Bibliografía del artículo
1. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J 20(1):1-8, 1958.
2. Richardson P, McKenna W, Bristow M y col. Report of the 1995 World Health Organization. International Society and Federation of Cardiology Task Force on the definition and Classification of Cardiomyopathies. Circulation 93:841-2, 1996.
3. Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 336(11):775-85, 1997.
4. Maron BJ, Moller JH, Seidman CE, y col. Impact of laboratory molecular diagnosis on contemporary diagnostic criteria for genetically transmitted cardiovascular diseases:hypertrophic cardiomyopathy, long-QT syndrome, and Marfan syndrome. A statement for healthcare professionals from the Councils on Clinical Cardiology, Cardiovascular Disease in the Young, and Basic Science, American Heart Association. Circulation 98(14):1460-71, 1998.
5. Maron BJ, Gross BW, Stark SI. Images in cardiovascular medicine. Extreme left ventricular hypertrophy. Circulation 92:2748, 1995.
6. Malik MS, Watkins H. The molecular genetics of hypertrophic cardiomyopathy. Curr Opin Cardiol 12(3):295-302, 1997.
7. Maron BJ. Risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Cardiol Rev 10(3):173-81, 2002.
8. Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet 363:1881-91, 2004.
9. Pelliccia A, Fagard R, Bjornstad HH, Anastassakis A, Arbustini E, Assanelli D y col. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J 26(14):1422-45, 2005.
10. Maron BJ, Olivotto I, Spirito P, y col. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 102(8),858-64, 2000.
11. Sherrid MV, Pearle G, Gunsburg DZ. Mechanism of benefit of negative inotropes in obstructive hypertrophic cardiomyopathy. Circulation 97(1):41-7, 1998.
12. Wigle ED, Rakowski H, Kimball BP, Williams WG. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation 92(7):1680-92, 1995.
13. Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 336(11):775-85, 1997.
14. Maron BJ, Spirito P, Shen WK, y col. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA 298(4):405-12, 2007.
15. Kuang SQ, Yu JD, Lu L, y col. Identification of a novel missense mutation in the cardiac beta-myosin heavy chain gene in a Chinese patient with sporadic hypertrophic cardiomyopathy. J Moll Cell Cardiol 28(9):1879-83, 1996.
16. Hagège AA, Dubourg O, Desnos M, y col. Familial hypertrophic cardiomyopathy. Cardiac ultrasonic abnormalities in genetically affected subjects without echocardiographic evidence of left ventricular hypertrophy. Eur Heart J 19(3):490-9, 1998.
17. Marian AJ, Roberts R. Recent advances in the molecular genetics of hypertrophic cardiomyopathy. Circulation 92(5):1336-47, 1995.
18. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4.111 subjects in the CARDIA Study. Circulation 92(4):785-9, 1995.
19. Niimura H, Bachinski LL, Sangwatanaroj S, y col. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 338(18):1248-57, 1998.
20. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4.111 subjects in the CARDIA Study. Circulation 92(4):785-9, 1995.
21. McKenna WJ, Monserrat L. Identification and treatment of patients with hypertrophic cardiomyopathy at risk of sudden death. Rev Esp Cardiol 53:123-130, 2000.
22. Elliott PM, Gimeno Blanes JR, Mahon NG, Poloniecki JD, McKenna WJ. Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet 357:420-24, 2001.
23. Jarcho JA, McKenna W, Pare JA, y col. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med 321(20):1372-8, 1989.
24. Geisterfer-Lowrance AA, Kass S, Tanigawa G, y col. A molecular basis for familial hypertrophic cardiomyopathy: a beta-cardiac myosin heavy chain gene missense mutation. Cell 62(5):999-1006, 1990.
25. Watkins H, MacRae C, Thierfelder L, Chou YH, Frenneaux M, McKenna W, Seidman JG, Seidman CE. A disease locus for familial hypertrophy cardiomyopathy maps to chromosome 1q3. Nat Genet 3(4):333-7, 1993.
26. Thierfelder L, MacRae C, Watkins H, y col. A familial hypertrophic cardiomyopathy locus maps to chromosome 15q2. Proc Natl Acad Sci USA 90(13):6270-4, 1993.
27. Carrier L, Hengstenberg C, Beckmann JS, y col. Mapping of a novel gene for familial hypertrophic cardiomyopathy to chromosome 11. Nat Genet 4(3):311-3, 1993.
28. Watkins H, Thierfelder L, Hwang DS, McKenna W, Seidman JG, Seidman CE. Sporadic hypertrophic cardiomyopathy due to de novo myosin mutations. J Clin Invest 90(5):1666-71, 1992.
29. Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman CE. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 77(5):701-12, 1994.
30. Osio A, Tan L, Chen SN, y col. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Cir Res 100(6):766-8, 2007.
31. Mohapatra B, Jimenez S, Lin JH, y col. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab 80(1-2):207-15, 2003.
32. Posch MG, Thiemann L, Tomasov P, y col. Sequence analysis of myozenin 2 in 438 European patients with familial hypertrophic cardiomyopathy. Med Sci Monit 14(7):CR372-4, 2008.
33. Geier C, Perrot A, Ozcelik C, y cols. Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation 107(10):1390-5, 2003.
34. Richard P, Charron P, Carrier L, y col. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107(17):2227-32, 2003.
35. Song L, Zou Y, Wang J, y col. Mutations profile in Chinese patients with hypertorphic cardiomyopathy. Clin Chim Acta 351(1-2):209-16, 2005.
36. Erdmann J, Daehmlow S, Wischke S, y col. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet 64(4):339-49, 2003.
37. Watkins H, Thierfelder L, Hwang DS, McKenna W, Seidman JG, Seidman CE. Sporadic hypertrophic cardiomyopathy due to de novo myosin mutations. J Clin Invest 90(5):1666-71, 1992.
38. Kimura A, Harada H, Park JE, y col. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 16(4):379-82, 1997.
39. Mogensen J, Klausen IC, Mogensen J, y col. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Inves 103:R39-43, 1999.
40. Burch M, Blair E. The inheritance of hypertrophic cardiomyopathy. Pediatr Cardiol 20(5):313-6, 1999.
41. Marian AJ, Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol 33(4):655-70, 2001.
42. McKenna WJ, Mogensen J, Elliott PM. Role of genotyping in risk factor assessment for sudden death in hypertrophic cardiomyopathy. J Am Coll Cardiol 39:2049-2051, 2002.
43. Elliott PM, Gimeno Blanes JR, Mahon NG, Poloniecki JD, McKenna WJ. Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet 357:420-24, 2001.
44. Rayment I, Holden HM, Sellers JR, Fananapazir L, Epstein ND. Structural interpretation of the mutations in the in the beta-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc Natl Acad Sci USA 92: 3864-8, 1995.
45. Roberts R, Sigwart U. New concepts in hypertrophic cardiomyopathies. Part I-Part II. Circulation 104:2113-6;2249-52, 2001.
46. Arad M, Seidman JG, Seidman CE. Phenotypic diversity in hypertrophic cardiomyopathy. Hum Mol Genet 11(20):2499-506, 2002.
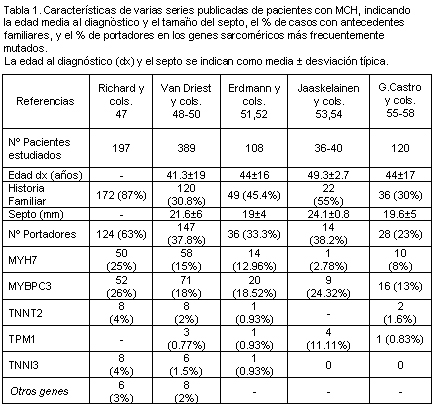
47. Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107(17):2227-32, 2003.
48. Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 108(4):445-51, 2003.
49. Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 44(3):602-10, 2004.
50. Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol 44(9):1903-10, 2004.
51. Erdmann J, Raible J, Maki-Abadi J, Hummel M, Hammann J, Wollnik B, Frantz E, Fleck E, Hetzer R, Regitz-Zagrosek V. Spectrum of clinical phenotypes and gene variants in cardiac myosin-binding protein C mutation carriers with hypertrophic cardiomyopathy. J Am Coll Cardiol 38(2):322-30, 2001.
52. Erdmann J, Daehmlow S, Wischke S, Senyuva M, Werner U, Raible J, Tanis N, Dyachenko S, Hummel M, Hetzer R, Regitz-Zagrosek V. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet 64(4):339-49, 2003.
53. Jääskeläinen P, Kuusisto J, Miettinen R Karkkainen P, Karkkainen S, Heikkinen S, Peltola P, Pihlajamaki J, Vauhkonen I, Laakso M. Mutations in the cardiac myosin-binding protein C gene are the predominant cause of familial hypertrophic cardiomyopathy in eastern Finland. J Mol Med 80(7):412-22, 2002.
54. Jääskeläinen P, Soranta M, Miettinen R, Saarinen L, Pihlajamaki J, Silvennoinen K, Tikanoja T, Laakso M, Kuusisto J. The cardiac beta-myosin heavy chain gene is not the predominant gene for hypertrophic cardiomyopathy in the Finnish population. J Am Coll Cardiol 32(6):1709-16, 1998.
55. García Castro M, Reguero JR, Batalla A, Díaz-Molina B, González P, Alvarez V, Cortina A, Cubero GI, Coto E. Hypertrophic cardiomyopathy: low frequency of mutations in the beta-myosin heavy chain (MYH7) and cardiac troponin T (TNNT2) genes among Spanish patients. Clin Chem 49:1279-85, 2003.
56. García Castro M, Reguero JR, Alvarez V, Batalla A, Soto MI, Albaladejo V, Coto E. Hypertrophic cardiomyopathy linked to homozygosity for a new mutation in the myosin-binding protein C gene (A627V) suggests a dosage effect. Int J Cardiol 102:501-7, 2005.
57. García Castro M, Reguero JR, Morís C, Alonso Montes C, Berrazueta JR, Sainz R, Alvarez V, Coto E. Prevalence and spectrum of mutations in the sarcomeric troponin T and I genes in a cohort of Spanish cardiac hypertrophy patients. Int J Cardiol 121:115-6, 2007.
58. García Castro M, Coto E, Reguero JR, Berrazueta JR, Alvarez V, Alonso B, Sainz R, Martín M, Morís C. Espectro mutacional de los genes sarcoméricos MYH7, MYBPC3, TNNT2, TNNI3 y TPM1 en pacientes con miocardiopatía hipertrófica. Rev Esp Cardiol, en prensa, 2008.