Bibliografía del artículo
Bibliografía del artículo
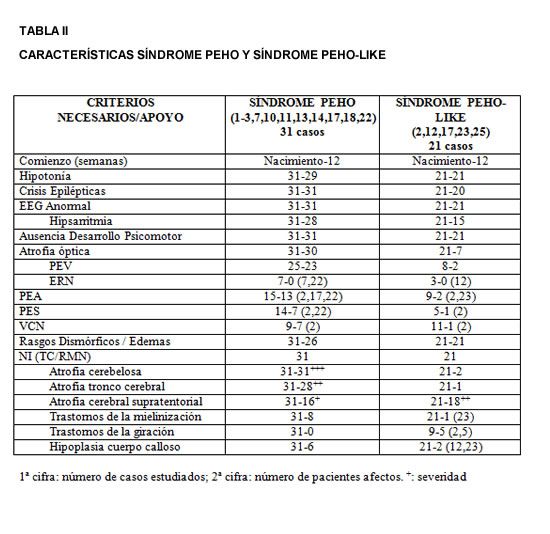
1. Salonen R, Somer M, Haltia M, Lorentz M, Noria R. Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO syndrome). Clin Genet 39:287-293, 1991.
2. Somer M. Diagnostic criteria and genetics of the PEHO syndrome. J Med Genet 30:932-936, 1993.
3. Somer M, Salonen O, Pihko H, Norio R. PEHO syndrome (progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy): Neuroradiologic findings. AJNR 14:861-867, 1993.
4. Somer M, Sainio K. Epilepsy and the electroencephalogram in progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (the PEHO syndrome). Epilepsia 34:72-731, 1993.
5. Haltia M, Somer M. Infantile cerebello-optic atrophy: Neuropathology of the progressive encephalopathy syndrome with edema, hypsarrhythmia and optic atrophy (the PEHO syndrome). Acta Neuropathol (Berl) 85:241-247, 1993.
6. Riikonen R, Somer M, Turpeinen U. Low insulin-like growth factor (IGF-1) in the cerebrospinal fluid of children with PEHO syndrome (progressive encephalopathy, hypsarrhythmia and optic atrophy) and cerebellar degeneration. Epilepsia 40:1642-1648, 1999. 7. Vanhatalo S, Riikonen R. Markedly elevated nitrate/nitrite levels in the cerebrospinal fluid of children with progressive encephalopathy with edema, hypsarrhythmia and optic atrophy (PEHO syndrome). Epilepsia 41:705-708, 2000.
8. Riikonen R. The PEHO syndrome. Brain Dev 23:765-769, 2001.
9. Caraballo R, Carignani M, Chamoles N, Fejerman N. PEHO syndrome in three non-Finnish patients. Pediatr Neurol 11:170, 1995.
10. Fujimoto S, Yokochi K, Nakano M, Wada Y. Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO syndrome) in two Japanese siblings. Neuropediatrics 26:270-272, 1995.
11. Shervell MI, Colangelo P, Treacy E, Polomeno RC, Rosenblatt B. Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO syndrome). Pediatr Neurol 15:337-339, 1996.
12. Chitty LS, Robb S, Berry C, Silver D, Baraitser M. PEHO or PEHO like syndrome?. Clin Dysmorphol 5:143-152, 1996.
13. Tanaka M, Tanaka Y, Hamano S, Nara T, Imai M. A case of PEHO (progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy) syndrome: Changes in clinical and neuroradiological findings. No To Hattatsu 29:488-493, 1997.
14. Hollody K, Kollar K. PEHO syndrome (progressive encephalopathy, edema, hypsarrhythmia, optic atrophy) [in Hungarian]. Orv Hetil 138:425-428, 1997.
15. Vanhatalo S, Somer M, Barth PG. Dutch patients with progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO) syndrome. Neuropediatrics 33:100-104, 2002.
16. Tekgul H, Tutuncuoglu S. Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO syndrome) in a Turkish child. Turk J Pediatr 42:246-249, 2000.
17. Field MJ, Grattan-Smith P, Piper SM, et al. PEHO and PEHO-like syndromes: Report of five Australian cases. Am J Genet 122A:6-12, 2003.
18. Nieto-Barrera M, Nieto-JimÉnez M, Diaz-FernÁndez F, Campaña-Marchal C, Candau FernÁndez-Mensaque R. Progressive encephalopathy with oedema, hypsarrhythmia, and optic atrophy (PEHO syndrome): A case report. Rev Neurol 36:1044-1046, 2003.
19. Klein A, Schmitt B, Boltshauser E. Progressive encephalopathy with edema, hypsarrhythmia and optic atrophy (PEHO) syndrome in a Swiss child. Eur J Paediatr Neurol 8:317-321, 2004.
20. Huisman TAGM, Klein A, Werner B, Straube T, Boltshauser E. Serial MR imaging, diffusion tensor imaging, and MR spectroscopic findings in a child with progressive encephalopathy, edema, hypsarrhythmia, and optic atrophy (PEHO). AJNR 27:1555-1558, 2006.
21. Sonmez G, Aydinöz S, Mutlu H, Ozturk E, Onur Sildiroglu H, Süleymanoglu S, et al. Serial MRI in a child with PEHO syndrome. J.Neurad 06.003:281-283, 2007.
22. Caraballo RH, Pozo AN, Gómez M, Semprino M. PEHO Syndrome: A Study of Five Argentinian Patients. Pediatric Neurology 44(4):259-264, 2010.
23. Logman C, Tolmie J, McWilliam R, MacLennan A. Cranial Magnetic resonance imaging mistakenly suggests prenatal ischaemia in PEHO-like syndrome. Clin Dysmorphol 12:133-136, 2003.
24. D'Arrigo S, Grazia BM, Faravelli F, Riva D, Pantaleoni C. Progressive encephalopathy with edema, hypsarrythmia, and optic nerve atrophy (PEHO)-like syndrome: what diagnostic characteristics are defining? J Child Neurol 20(5):454-456, 2005.
25. Goizet C, Espil-Taris C, Husson M, Chateil JF, Pedespan JM, Lacombe D. A patient with hydranencephaly and PEHO-like dysmorphic features. Ann Genet 46(1):25-28, 2003.
26. Barber JCK, Maloney VK, Bewes B, Wakeling E. Deletions of 2q14 that include the homeobox engrailed 1 (EN1) transcription factor are compatible with a normal phenotype. Eur J Hum Genet 14:739-743, 2006.
27. Aicardi J, Chevrie JJ, Rousselie F. Le syndrome spasmes en flexion agénésie calleuse, anomalies chorioretiniennes. Arch Fr Pédiatr 26:1103-1120, 1969.
28. Norman RM. Primary degeneration of the granular layer of the cerebellum: an unusual form of familial cerebellar atrophy occurring in early life. Brain 63:365-379, 1940.