Bibliografía del artículo
Bibliografía del artículo
1. Bush A. Paediatric interstitial lung disease: not just kid's stuff. Eur Respir J 24:521-3, 2004.
2. Fan LL, Deterding RR, Langston C. Pediatric interstitial lung disease revisited. Pediatric Pulmonol 38:369-78, 2004.
3. Clement A, Nathan N, Epaud R, Fauroux B, Corvol H. Interstitial lung diseases in children. Orphanet J Rare Dis 5:22, 2010.
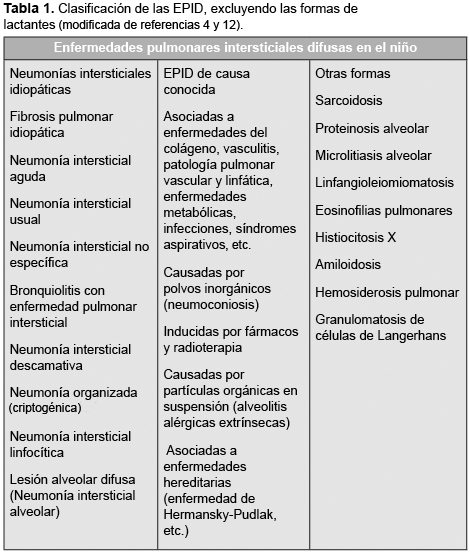
4. Clement A, and committee members. Task force on chronic interstitial lung disease in immunocompetent children. Eur Respir J 24:686-97, 2004.
5. Barbato A, Panizzolo C. Chronic interstitial lung disease in children. Paediatric Respir Rev 1:172-8, 2000.
6. Clement A, Eber E. Interstitial lung diseases in infants and children. Eur Respir J 31:658-66, 2008.
7. Cruzado V, Tolín M, Berroya A, Navarro N, Rodríguez-Cimadevilla J, Salcedo A. Enfermedades pulmonares intersticiales difusas en el paciente pediátrico. Rev Esp Pediatr 64:419-25, 2008.
8. Paiva MA, Amaral SM. Chronic interstitial lung diseases in children. J Bras Pneumol 35:792-803, 2009.
9. Deterding RR, Fan LL, Morton R, Hay TC, Langston C. Persistent tachypnea of infancy (PTI)- a new entity. Pediatric Pulmonol 23:72-3, 2001.
10. Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG, Dell S, Fan LL, Hamvas A, Hilman BC, Langston C, Nogee LM, Redding GJ; American Thoracic Society Committee on Childhood Interstitial Lung Disease (chILD) and the chILD Research Network. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med 188:376-94, 2013.
11. Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, Brody AS, Nogee LM, Trapnell BC, Langston C, et al.; Pathology Cooperative Group; ChILD Research Co-operative. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med 176:1120-1128, 2007.
12. Xaubet A, Ancochea J, Blanquer R, Montero C, Morell F. Diagnóstico y tratamiento de las enfermedades pulmonares intersticiales difusas. Arch Bronconeumol 39:580-600, 2003.
13. Miller JD, Urschel JD, Cox G, Olak J, Young JE, Kay JM, McDonald E. A randomized, controlled trial comparing thoracoscopy and limited thoracotomy for lung biopsy in interstitial lung disease. Ann Thorac Surg 70:1647-50, 2000.
14. Hilman BC, Amaro-Galvez R. Diagnosis of interstitial lung disease in children. Paediatr Respir Rev 5:101-7, 2004.
15. Lettieri CJ, Veerappan GR, Helman DL, Mulligan CR, Shorr AF. Outcomes and safety of surgical lung biopsy for interstitial lung disease. Chest 127:1600-5, 2005.
16. Lee YC, Wu CT, Hsu HH, Huang PM, Chang YL. Surgical lung biopsy for diffuse pulmonary disease: experience of 196 patients. J Thorac Cardiovasc Surg 129:984-90, 2005.
17. Dinwiddie R. Treatment of interstitial lung disease in children. Paediatr Respir Rev 5:108-15, 2004.
18. Bush A. Pediatric interstitial lung disease. Breathe 2:17-29, 2005.
19. American Thoracic Society, European Respiratory Society, Join Statement of the American Thoracic Society and the European Respiratory Society. American Thoracic Society/ European Respiratory Society international multidisciplinary consensus classification of the idiopatic interstitial pneumonias. Am J Respir Crit Care Med 165:277-304, 2002.