RESULTADOS ACTUALIZADOS DEL RASTREO MASIVO PARA LA DETECCION DE ESFEROCITOSIS HEREDITARIA EN LUXEMBURGO. PREVALENCIA, FISIOPATOLOGÍA Y CONSECUENCIAS
(especial para SIIC © Derechos reservados)
Coautores
Jerzy Janecki* Nathalie Coulon** Martine Thoma*** Pierre Kutter****
Prof MD, Institue of Biocybernetics and Bionedical Enineering ? Polish Academjy of Sciences ? 4 Kkssieçcia Troidena St ? Warszawa (Poland)*
Senior Med Technologist, Laboratoires réunis Kutter-Lieners Hastert ? PO Box 11 ? L6101 Junglinster (Grand Duchy of Luxembourg)**
Msc, Laboratoires réunis Kutter-Lieners Hastert ? PO Box 11 ? L6101 Junglinster (Grand Duchy of Luxembourg)***
MD, Laboratoires réunis Kutter-Lieners Hastert ? PO Box 11 ? L6101 Junglinster (Grand Duchy of Luxembourg)****
Recepción del artículo: 28 de noviembre, 2003
Aprobación: 0 de , 0000
 Conclusión breve
El estudio realizado en Luxemburgo demuestra que la prevalencia de esferocitosis hereditaria es mucho más alta que la considerada hasta ahora.
Resumen
Los estudios hematológicos automáticos con difracción doble con rayo láser mediante la transformación artificial de glóbulos rojos esféricos determinan el volumen y la concentración de hemoglobina de un gran número de células, incluso el porcentaje de elementos aberrantes. La identidad de los eritrocitos hipercrómicos y de los esferocitos, así como su proporción normal, se han establecido previamente. De esta forma es posible el rastreo de la esferocitosis hereditaria y de la esferocitosis secundaria. Al aplicar ciertos criterios –permanencia de mayor porcentaje de esferocitos, historia familiar, hallazgos bioquímicos de hemólisis y los datos clínicos– determinamos una prevalencia de esferocitosis hereditaria (EH) generalmente asintomática de 1:300 en hombres y de 1:750 en mujeres, valores mucho más elevados que los admitidos hasta ahora de 1:5 000. Las manifestaciones clínicas como ictericia, esplenomegalia, anemia, aplasia y cálculos biliares, que hacen sospechar el diagnóstico de esferocitosis son, en realidad, raras. Este hecho explica la discrepancia entre estos valores. Debido a que la EH es un defecto hemolítico no fue inesperada la frecuente sobrecarga de hierro. La elevada asociación de EH con diabetes y sobrecarga de hierro sugiere daño pancréatico por la acumulación, tal como se observa en la hemocromatosis genética. El médico y el enfermo deben informarse acerca de la presencia de EH para evitar la interpretación errónea de las manifestaciones patológicas. Se recomienda el monitoreo de glucemia y ferritina.
Palabras clave
Esferocitosis hereditaria, esferocitosis secundaria, hipercromía de glóbulos rojos
Clasificación en siicsalud
Conclusión breve
El estudio realizado en Luxemburgo demuestra que la prevalencia de esferocitosis hereditaria es mucho más alta que la considerada hasta ahora.
Resumen
Los estudios hematológicos automáticos con difracción doble con rayo láser mediante la transformación artificial de glóbulos rojos esféricos determinan el volumen y la concentración de hemoglobina de un gran número de células, incluso el porcentaje de elementos aberrantes. La identidad de los eritrocitos hipercrómicos y de los esferocitos, así como su proporción normal, se han establecido previamente. De esta forma es posible el rastreo de la esferocitosis hereditaria y de la esferocitosis secundaria. Al aplicar ciertos criterios –permanencia de mayor porcentaje de esferocitos, historia familiar, hallazgos bioquímicos de hemólisis y los datos clínicos– determinamos una prevalencia de esferocitosis hereditaria (EH) generalmente asintomática de 1:300 en hombres y de 1:750 en mujeres, valores mucho más elevados que los admitidos hasta ahora de 1:5 000. Las manifestaciones clínicas como ictericia, esplenomegalia, anemia, aplasia y cálculos biliares, que hacen sospechar el diagnóstico de esferocitosis son, en realidad, raras. Este hecho explica la discrepancia entre estos valores. Debido a que la EH es un defecto hemolítico no fue inesperada la frecuente sobrecarga de hierro. La elevada asociación de EH con diabetes y sobrecarga de hierro sugiere daño pancréatico por la acumulación, tal como se observa en la hemocromatosis genética. El médico y el enfermo deben informarse acerca de la presencia de EH para evitar la interpretación errónea de las manifestaciones patológicas. Se recomienda el monitoreo de glucemia y ferritina.
Palabras clave
Esferocitosis hereditaria, esferocitosis secundaria, hipercromía de glóbulos rojos
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/20030
Especialidades
 Principal: Hematología,
Principal: Hematología,
Relacionadas: Diagnóstico por Laboratorio, Epidemiología, Genética Humana, Medicina Interna,
Enviar correspondencia a:
Dolphe Kutter. PO Box 11 L 6101 Junglinste Grand Duchy of Luxembourg
UPDATED RESULTS OF MASS SCREENING FOR HEREDITARY SPHEROCYTOSIS IN LUXEMBOURG. PREVALENCE, PHYSIOPATHOLOGY, CONSEQUENCES
Abstract
Hematological automates using double beam laser diffraction by artificially spherized red blood cells determine both volume and hemoglobin concentration of a very large number of cells, even indicating percentages of aberrant elements. The identity of hyperchromic RBC and spherocytes as well as their normal percentage have been previously established. Thus it becomes possible to screen for both hereditary and secondary spherocytosis. Applying criteria such as permanence of an increased percentage of spherocytes, family history, biochemical symptoms of hemolysis and clinical data, we claim a prevalence of generally asymptomatic hereditary spherocytosis (HS) of 1:300 men and 1:750 women, values much higher than the hitherto admitted 1:5 000. Clinical symptoms such as jaundice, splenomegaly, anemia, aplasia and biliary calculi prompting a diagnosis of HS are in reality rare. This explains the discrepancy between these values. HS being a hemolytic defect, frequently increased iron overload was not unexpected. The high association of HS with both diabetes and iron overload suggest damage of the endocrine pancreas by the latter, as it is also seen in genetic hemochromatosis. Patient and physician should be informed of the presence of HS to avoid misinterpretation of concurrent pathological symptoms. Monitoring of blood glucose and ferritin is recommended.
Key words
Esferocitosis hereditaria, esferocitosis secundaria, hipercromía de glóbulos rojos
RESULTADOS ACTUALIZADOS DEL RASTREO MASIVO PARA LA DETECCION DE ESFEROCITOSIS HEREDITARIA EN LUXEMBURGO. PREVALENCIA, FISIOPATOLOGÍA Y CONSECUENCIAS
(especial para SIIC © Derechos reservados)
Artículo completo
Introducción
La esferocitosis hereditaria (EH) fue descrita por Vanlair y Masius en 18711 como microcitemia y no en la forma referida generalmente por Minkowski2 y Chauffard.3 La EH es un trastorno hemolítico familiar caracterizado por anemia, ictericia intermitente, esplenomegalia y respuesta a la esplenectomía. Un estudio de Haden, de 1934,4 llamó la atención sobre una probable anormalidad estructural de la membrana de los glóbulos rojos (GR) como causa de la hemólisis. Estudios más extensos mostraron que la EH es un trastorno hemolítico originado por deficiencias genéticas de una o más proteínas del citoesqueleto de los GR, α y β espectrina, proteína de banda 4.2, anquirina y proteína de banda 3.
Las mutaciones de los genes que codifican la síntesis de dichas proteínas ocasionan en la mayoría de los casos su deficiencia o su ausencia absoluta; la alteración genera consecuencias más o menos marcadas en la estructura de la membrana de los GR. Las deficiencias de estas proteínas desestabilizan la doble capa lipídica de la membrana de los GR, con pérdida de área de superficie de membrana por la formación de vesículas, deshidratación, transformación a células esféricas, mayor concentración de hemoglobina, aumento de la rigidez celular y de la fragilidad y, finalmente, hemólisis. Los microesferocitos que se observan en el estudio microscópico de rutina en combinación con síntomas clínicos evidentes fueron, durante largo tiempo, las bases para realizar el diagnóstico.
La EH se transmite más frecuentemente en forma autosómica dominante. Los individuos afectados son heterocigotas ya que la alteración homocigota se considera incompatible con la vida. Se estima que en el 25% de los casos hay transmisión recesiva, sospechada cuando padres aparentemente normales tienen más de un hijo con la deficiencia. Las mutaciones espontáneas de novo parecen posibles.
Según el tipo de proteína y la importancia de la deficiencia, la EH ocurre con marcada heterogeneidad clínica, desde una patología asintomática hasta anemia hemolítica fatal y aplasia que pone en peligro la vida.
La microscopia, los síntomas clínicos y los estudios familiares motivaron la creencia de que la EH era un trastorno bastante raro que ocurría con una frecuencia de 1:5 000 individuos, esencialmente oriundos del norte de Europa.4
El microesferocito se caracteriza por fragilidad osmótica anormal in vitro. Los grupos que rastrearon esta mayor fragilidad de GR en dadores normales con diversas técnicas como el pink test, prueba de lisis con glicerol y prueba de criólisis,5-9 sugieren la posibilidad de una prevalencia mucho más alta. La separación y cuantificación de las proteínas de membrana con electroforesis en gel de poliacrilamida (PAGE) permite la caracterización precisa de la alteración. Sin embargo, estos procedimientos sólo se pueden realizar en centros especializados.
El rastreo masivo de la esferocitosis, sea cual fuere el origen, se hizo posible en los estudios hematológicos de rutina por el uso de varios tipos de analizadores automáticos. La medición por citometría de flujo de la difracción de rayo láser de doble ángulo de GR artificialmente transformados en esferocitos permite determinar el volumen y la densidad de los GR; en otras palabras, puede establecer la concentración de hemoglobina de aproximadamente 50 000 células. Mediante esta técnica, el analizador hematológico Advia® 120 (Bayer Inc., Terrytown, EE.UU.) es capaz de diferenciar y cuantificar diversas anormalidades de volumen y concentración de una determinada población en el denominado eritrograma (Figura 1a), incluso el porcentaje exacto de eritrocitos aberrantes. La figura 1b muestra un eritrograma típico de un paciente con EH con un alto porcentaje de GR hipercrómicos. Mediante criólisis para determinar mayor fragilidad celular pudimos establecer que estos GR hipercrómicos son en realidad esferocitos y que su incremento se asocia con mayor hemólisis.10,11 Por ello el aumento marcado del porcentaje de hipercromía se transforma en una manifestación principal de la EH. Los GR hipercrómicos han generado una enorme controversia con los hematólogos de mayor edad que consideran que es imposible que el GR se sobrecargue.12,13 Algunas investigaciones, entre ellas, la de Mohandas y col.14 y la de Ialongo y col.15 demuestran la génesis de esferocitos hipercrómicos por pérdida de membrana en forma de vesículas por ensamblado anormal de las proteínas de membrana. Como consecuencia hay deshidratación y descenso del volumen. En la EH este trastorno tiene lugar en el estadio de reticulocito macrocítico y esto explica por qué los esferocitos finales son en su mayoría normocíticos. El uso de este analizador muestra porcentajes bajos de eritrocitos/esferocitos hipercrómicos (%Hiper) en la gran mayoría de los enfermos que se estudian en forma rutinaria. La proporción de esferocitos sin embargo, nunca es cero. Probablemente se formen durante el proceso normal de envejecimiento de los GR. Por ello fue necesario actualizar los hallazgos previos sobre el %Hiper normal.

Figura 1. Eritrogramas: a) normal (2.3 %Hiper); b) caso típico de esferocitosis hereditaria (38.9 %Hiper).
A partir de estos datos determinamos criterios mínimos para establecer esferocitosis significativa y seleccionar casos con elevada probabilidad de EH. En estos sujetos tratamos de obtener la mayor información personal, clínica y de laboratorio, no sólo de establecer la prevalencia de EH sino también las anomalías frecuentemente asociadas. En los restantes casos se consideraron causas de esferocitosis secundaria.
Los pacientes con EH confirmada en centros especializados y por historia familiar inequívoca permitieron evaluar las variaciones intraindividuales del síntoma "mayor %Hiper" y determinar los valores más bajos, aún compatibles con el diagnóstico de EH.
Métodos y población investigada
El estudio rutinario hematológico se realizó con un analizador automático Advia® 120 (Bayer Inc., Terrytown, EE.UU.). El control de calidad especial de %Hiper se obtuvo por comparación de los valores diarios promedio de este parámetro. La prueba de criólisis descrita por Rosas-Romero y col.8 se utilizó para evaluar la fragilidad de los GR. Otros parámetros relevantes como ferritina, hierro sérico, saturación de transferrina, bilirrubina libre y total, haptoglobina, enzimas hepáticas, glucemia y hemoglobina glucosilada (Hb1Ac) se determinaron con métodos convencionales en uso en el laboratorio, bajo controles permanentes de calidad, internos y externos.
Se necesita una variable preliminar para caracterizar la población de estudio: nuestro instituto es un laboratorio privado de rutina que realiza exámenes que indica el médico y que son reintegrados por el seguro social. Esto significa que nuestras investigaciones están limitadas a las prescritas, hecho que explica la gran cantidad de casos con datos incompletos. Las enfermedades graves son raras. Nuestros enfermos son en su mayoría ambulatorios, generalmente derivados para estudios de rutina o por patologías menores al laboratorio principal o a alguno de los centros distribuidos en todo el país. Nuestros extraccionistas sólo obtienen algo de información clínica y las solicitudes de los médicos normalmente carecen de ella. Nuestros resultados, por lo tanto, no pueden ser tomados como valores normales de referencia. No obstante, los consideramos cercanos a los habituales.
Hemos evaluado aproximadamente 75 000 pacientes de rutina, en su mayoría adultos, durante 33 meses desde 2001 a 2003. La relación hombre/mujer es de 0.59. En ocho pacientes el diagnóstico de EH se confirmó en instituciones especializadas. Cuatro de ellos se presentaron junto con 5 familiares directos con EH evidente (1 hermano; 3 hijos y el padre).
Criterios para el diagnóstico de EH
El diagnóstico definitivo de EH sólo es posible mediante la cuantificación de las diferentes proteínas del citoesqueleto, un procedimiento que no está al alcance de los laboratorios de rutina. Sobre la base de los datos de enfermos con EH conocida y de estudios previos16,17 establecimos los siguientes criterios:
- El síntoma principal es el incremento del %Hiper durante al menos un año y con un valor de 9.5% o más en al menos una oportunidad; eventualmente, valores tan bajos como 3.5% pero ningún valor en el área normal. Una detección aislada de %Hiper no es prueba suficiente para el diagnóstico de EH.
- La existencia de miembros familiares directos (padres, hijos, hermanos) con aumento del %Hiper (3.5% o más) representa un argumento adicional de valor en el diagnóstico de EH tanto para el enfermo como para sus parientes.
- Otros síntomas de hemólisis y sobrecarga de hierro (reticulocitos altos, aumento de la bilirrubina libre, descenso de la haptoglobina, incremento de la ferritina o de la saturación de transferrina, urobilinogenuria) avalan el diagnóstico de EH.
- Los síntomas clínicos principales son ictericia (eventualmente intermitente), rara vez litiasis biliar.
Los siguientes valores se consideran límites normales de ferritina de acuerdo con la Sociedad Americana de Hemocromatosis.

Resultados
Se actualizaron los valores normales del %Hiper en dos artículos previos con un programa computarizado especial elaborado por uno de nosotros (JJ) para la delimitación de las subpoblaciones gaussianas.18 Con este equipo se analizaron los valores del %Hiper de 7 820 pacientes de rutina. Tal como se observa en la tabla I obtuvimos 3 subpoblaciones. Los valores clave son prácticamente idénticos a los que se obtuvieron en estudios más pequeños:10,17 en la gran mayoría, los valores oscilaron entre 0 y 3.4%, lo que representa la población normal. Consideramos valores moderadamente elevados entre 3.5% y 9.4% mientras que 9.5% o más se consideró aumento marcado. La superposición sustancial de las dos últimas curvas gaussianas genera una separación débil de estas dos subpoblaciones.

Tabla I. Distribución del porcentaje de hipercromía (%Hiper) en un grupo de 7.820 individuos no seleccionados.7
En 7 casos con EH conocida pudo seguirse la evolución. Los resultados se muestran en la figura 2; confirman los criterios mencionados previamente.
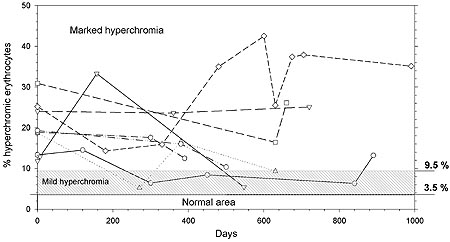
Figura 2. Evolución de %Hiper en 7 pacientes con esferocitosis hereditaria establecida.
Entre 75 000 individuos estudiados encontramos 319 pacientes con valores de %Hiper de 9.5% o más. Según los criterios mencionados, en 165 se realizó directamente el diagnóstico de EH, en 20 por la investigación familiar. Su distribución y prevalencia por género se muestra en la tabla II.

Tabla II. Casos de esferocitosis hereditaria (EH) entre los 75 000 individuos rastreados.
Tal como se mencionó observamos fluctuaciones importantes del %Hiper en muchos de los casos detectados, incluso períodos prolongados de hipercromía leve, fenómeno que sugiere que la EH evoluciona con fases de actividad separadas por períodos más o menos inactivos. Sin embargo, consideramos que la aparición de valores normales de %Hiper excluye el diagnóstico de EH. El origen étnico de los 185 casos de EH se muestra en la tabla III. La tabla IV brinda una visión general de los datos adicionales de laboratorio disponibles en estos pacientes. Los síntomas clínicos son infrecuentes. Cinco hombres presentaron esplenomegalia y dos de ellos fueron sometidos a esplenectomía. Otros dos enfermos fueron sometidos a cirugía por litiasis biliar; lamentablemente no su pudo realizar el estudio de los cálculos eliminados. Tres pacientes tuvieron ictericia leve intermitente.

Tabla III. Distribución étnica de 185 casos de EH comparada con la población general del Gran Ducado de Luxemburgo.

Tabla IV. Datos disponibles de laboratorio en 185 casos de esferocitosis hereditaria. Ningún caso de aplasia.
Dado que la EH es un trastorno hemolítico hicimos hincapié en el metabolismo del hierro. En 137 enfermos se dispuso de información sobre los valores de ferritina o saturación de transferrina. El estado civil se muestra en la tabla V.

Tabla V. Sobrecarga de hierro mostrada por el incremento de hierro en 185 pacientes con esferocitosis hereditaria. Véase el texto para los criterios de ferritina normal.
Desde el principio de nuestra investigación notamos la incidencia alta de diabetes no sólo en los pacientes con EH confirmada sino también entre los individuos con hipercromía importante en quienes el diagnóstico está pendiente. En la tabla VI se muestra la incidencia de diabetes o intolerancia a la glucosa en 167 pacientes con EH en quienes se dispone de información acerca del metabolismo de la glucosa.

Tabla VI. Diabetes o intolerancia a la glucosa entre 167 pacientes con esferocitosis hereditaria e información disponible del metabolismo de la glucosa.
En base a la sospecha de asociación entre EH, diabetes y sobrecarga de hierro19,20 seleccionamos los casos de EH con datos de glucemia y del metabolismo férrico (tabla VII). Aún no extendimos nuestra investigación a pacientes con hipercromía leve permanente.

Tabla VII. Incidencia de sobrecarga de hierro en 47 pacientes con esferocitosis hereditaria y diabetes o intolerancia a la glucosa.
La marcada hipercromía intermitente entre valores normales sugiere esferocitosis secundaria. En 5 casos, el incremento aparece en el tercer trimestre de la gestación. En seis casos, la hipercromía pudo explicarse por quimioterapia antitumoral. En tres pacientes la hipercromía se constata junto con un descenso muy notorio de la osmolaridad en plasma por tratamiento excesivo con diuréticos. Una única medición de hipercromía marcada pudo atribuirse a deficiencia de G6PDH. Se diagnosticó un caso con síndrome HELLP en la fase inicial por el incremento del %Hiper y enzimas hepáticas en combinación con descenso de trombocitos. En estos 10 casos se excluyó EH.
En 154 enfermos, los datos son insuficientes para establecer o eliminar el diagnóstico de EH. Treinta de ellos tienen diabetes, 17 en asociación con sobrecarga de hierro, 13 con estado de hierro normal o desconocido. Cuarenta y cinco sujetos tienen sobrecarga de hierro, 34 sin diabetes y 11 sin datos acerca del metabolismo de la glucosa.
Discusión
Nuestros resultados demuestran claramente que la EH es mucho más frecuente que lo que se consideraba hasta ahora (1:5 000). La gran mayoría de nuestros casos se detectaron sólo mediante el hallazgo de marcada hipercromía de los GR que mostró el analizador hematológico. La anemia no parece ser un síntoma importante en EH. La prueba de criólisis, aunque es un indicador extremadamente sensible de esferocitosis, sólo se solicita en forma excepcional. El estudio hematológico de rutina no incluye el recuento de reticulocitos, un parámetro con sensibilidad moderada. El descenso de la haptoglobina –también solicitada muy rara vez– es inesperado ya que la EH es un proceso hemolítico extravascular. La lisis parcial intravascular de los GR por fragilidad excesiva podría explicar dicha controversia. En la práctica, el incremento moderado de la bilirrubina se atribuye por lo general a deficiencia de UDP-glucuronosil-transferasa (síndrome de Gilbert) y se deja de lado sin más investigación. Es excepcional que las pruebas de laboratorio de rutina sugieran EH. Este hecho, en combinación con los síntomas clínicos raros e inespecíficos, explica por qué la mayoría de nuestros casos podrían haber permanecido sin detección si no se determinaba la concentración de hemoglobina de los GR y el porcentaje de células hipercrómicas. También explica la discrepancia entre nuestras cifras y la prevalencia generalmente aceptada de 1:5 000.
La distribución étnica de nuestros casos de EH es homogénea en toda Europa y excluye la prevalencia más alta entre los habitantes del norte de Europa tal como lo describió Minkowski.2 Una proporción de casos de Italia y de la Península Ibérica sólo se explica por el alto número de pacientes de ese origen en la población de Luxemburgo (tabla II).
Con la actualización de los datos de laboratorio de nuestros pacientes con EH más o menos confirmamos nuestros resultados anteriores,11 especialmente la elevada incidencia de sobrecarga de hierro en varones. Esto se explica fácilmente por la corta vida media de los esferocitos y la mayor acumulación de hierro liberado. En mujeres, este fenómeno se suprime parcialmente por las pérdidas menstruales. Beutler y col.21 concluyen en que el efecto de la sobrecarga de hierro en la hemocromatosis hereditaria es bajo; encuentran prácticamente los mismos síntomas en una amplia población portadora homocigota de la mutación C282Y y el grupo control comparable. Aun así se considera que la sobrecarga de hierro –sea cual fuere el origen– es tóxica para muchos órganos y causa daño al hígado, riñones y sistema inmune y origina diabetes y anormalidades hormonales,2,22,23 fue reconocida por muchos autores como factor casual en enfermedad neoplásica y en infección bacteriana y viral.24-28 La relevancia de la sobrecarga de hierro en arteriosclerosis e infarto de miocardio es controvertida. Varios grupos consideran que existe una clara relación entre ambos factores.29-31 Más aun, Passa y col.32 recomiendan terapia preventiva con flebotomías. Otros grupos no encontraron asociación entre ambas patologías.33,34 Debido a que generalmente desconocemos el estado cardiológico de nuestros enfermos no podemos participar en esta discusión.
Nuestros datos muestran asociación muy frecuente entre EH y diabetes, lo cual confirma nuestras observaciones anteriores (tabla IV). Nuestros experimentos y los resultados en diabetes mal controlada contradicen un efecto simple a corto plazo de hiperosmolaridad mediada por glucosa como causa de mayor %Hiper.20 La glucosilación exagerada de las proteínas de membrana es una hipótesis obvia. Varias publicaciones demuestran modificaciones en la membrana de los eritrocitos por glucosilación y mayor hemólisis en diabéticos, con énfasis en la mayor rigidez.35-38 Przybylska, Bryszewska y Chapman claramente involucraron una ruptura en la bicapa de la membrana,39 similar a la que se observa en el proceso de la esferocitosis.15 Esto significa que la glucemia elevada puede asociarse con esferocitosis adquirida, secundaria y con mayor hemólisis por glucosilación de la estructura de la membrana. No podemos explicar por qué esto no sucede en todos los sujetos con mal control de la diabetes. Es posible que este efecto se limite principalmente a individuos con defectos subyacentes –eventualmente mínimos y ocultos– de la membrana. Aunque no excluimos este tipo de esferocitosis adquirida, creemos en una acción directa de la sobrecarga de hierro sobre el páncreas endocrino, tal como ocurre en la "diabetes bronce", como consecuencia de una asociación entre hemocromatosis genética y diabetes. Nuestros casos de EH con información acerca del metabolismo de la glucemia y del estado férrico (tabla VII) parecen confirmar esta hipótesis, también presentada por otros autores en relación con hemocromatosis por mutaciones genéticas.40-42 Aun se discute la posible prevención de la diabetes por reducción de la sobrecarga de hierro. Parece razonable aplicar el mismo argumento en EH.
Esto motiva un interrogante importante: ¿qué debemos decir a los pacientes en quienes se establece el diagnóstico de EH Consideramos que el enfermo y su médico deben conocer el diagnóstico, aun cuando la patología sea completamente asintomática. El paciente debe saber que en la gran mayoría de los casos la anomalía no se asocia con síntomas importantes. El riesgo de sobrecarga de hierro y diabetes debería, sin embargo, discutirse; se recomienda el monitoreo regular de la ferritina y de la glucosa en sangre. El significado de los síntomas clínicos debería conocerse, particularmente en relación con la ictericia intermitente. Hemos visto un paciente con EH tratado por ictericia obstructiva durante una fase de actividad de su EH. En cambio, la elevación de la bilirrubina total e ictericia no deben atribuirse automáticamente a EH y hemólisis. La esplenomegalia y la litiasis biliar pueden ser causa de dolor abdominal. La palpación del bazo y la ecografía pueden esclarecer la causa de los síntomas. Debe tenerse en cuenta la posibilidad de una crisis aplásica en pacientes con anemia excesiva.
La hipercromía aumenta regularmente durante el embarazo, incluso a valores sugestivos de hipercromía marcada y en presencia de valores normales durante el primer trimestre. Los incrementos extremos deben motivar un recuento de plaquetas y la determinación de las enzimas hepáticas para excluir o confirmar el diagnóstico del síndrome HELLP.
El casamiento de portadores de EH clásica se asocia con el riesgo de descendencia homocigota no viable. El rastreo de personas que se casan o viven en pareja permite descubrir dicha coincidencia y el riesgo. Puede brindar una explicación para casos de abortos repetidos.
La interpretación de la hipercromía leve permanente es difícil. Estamos de acuerdo con Conway y col.43 quienes consideran que estos enfermos presentan deficiencias leves parciales o heterocigotas de las proteínas del esqueleto. Este grupo sin duda alberga casos de EH verdadera en su fase latente. La prevalencia de EH podría por ende ser mayor a la que se consideró anteriormente.
Conclusiones
La difracción de rayo láser de doble ángulo de los GR artificialmente transformados a esferocitos brinda información cuantitativa del volumen y de la concentración de hemoglobina e indica los porcentajes de células aberrantes. Se demostró que los GR hipercrómicos corresponden a esferocitos. Esto permite un rastreo libre de costos para EH en los estudios hematológicos rutinarios con equipos automáticos sobre la base de este principio. La prevalencia de EH se estimó en aproximadamente 1:300 en hombres y 1:750 en mujeres. La discrepancia con la prevalencia considerada hasta ahora de 1:5 000 se explica por el hecho de que la mayoría de los casos no tienen síntomas clínicos. Los datos adicionales de laboratorio disponibles muestran, sin embargo, una elevada frecuencia de sobrecarga de hierro, por la menor vida media de los GR anormales. El efecto tóxico de la sobrecarga férrica podría ser responsable de daño endocrino al páncreas y de la alta incidencia de diabetes. Tanto los pacientes con EH como los médicos involucrados en su atención deberían ser informados acerca de la presencia de la anomalía, que podría ser la explicación de otras patologías simultáneas y motivar asesoramiento genético.
Los autores no manifiestan conflictos
Bibliografía del artículo
- Vanlair CF, Masius JB. De la microcythémie. Bull R Acad Méd Belg 1871;5:515-613.
- Minkowski O. Über eine hereditäre, unter dem Bilde eines chronischen Ikterus mit Urobilinurie, Splenomegalie und Nierensiderosis verlaufende Affektion. Verhandlungen des Kongresses für innere Medizin, 1900; 18:316-323.
- Chauffard M A. Pathogénie de l\'ictère congénital de l\'adulte. La semaine médicale 1907; 27:25-29.
- cited by Lee RG in: Wintrobe\'s Clinical Hematology. 10th edition. Lippingcott, Williams and Wilkins 1999 (Baltimore).
- Godal HC, Heist H. High prevalence of increased osmotic fragility among Norwegian blood donors. Scand J Haematol 1981; 27:30-33.
- Rudolphi O, Hornsten P. Hereditary spherocytosis is more common than expected. Lakatidningen 1996; 93:1745-1748.
- Eber SW, Pekun A, Neufeldt A, Schroter W. Prevalence of increased osmotic fragility of erythrocytes in German blood donors – screening using a modified glycerol lysis test. Ann. Hematol 1992; 64:88-91.
- Rosas-Romero R, Poo JL, Robles JA, Uriostegui A, Vargas F, Majluf-Cruz A. Usefulness of the cryohemolysis test in the diagnosis of hereditary spherocytosis. Archives of Medical Research 1997; 28;247-251.
- Mittler U, Radig K, Kluba U, Aumann V, Röppnack R. Erfahrungen mit dem Glyzerol- Lyse-Test im sauren Milieu. Kinderärtzl Praxis 1993; 61:219-222.
- Kutter D, Coulon N, Stirn F, Thoma M, Janecki J. Demonstration and quantification of "hyperchromic" erythrocytes by haematological analysers. Application to screening for hereditary and acquired spherocytosis, Clin Lab 2002; 48:163-170.
- Kutter D, Mijanovic-Lasic D, Kremer A, Routinemässige Erfassung einer extravasalen Hämolyse durch Erfassung von Hyperchromie und Sphärozytose. Berichte der ÖGKC 2002; 25:5-10.
- Wintrobe M M. The size and hemoglobin content of the erythrocyte. J Lab Clin Med 1932; 17:899-904.
- Wintrobe M M. Anaemia : Classification and treatment on the basis of differences in the average volume and hemoglobin content of the red corpuscules. Arch Intern Med 1934; 54:256-261.
- Mohandas N, Johnson A, Wyatt J, Croisille L, Reeves J, Tycko D, Groner W. Automated quantitation of cell densitiy distribution and hyperdense cell fraction in RBC disorders. Blood 1989; 74 : 442-447.
- Ialongo PL, Vignetti M, Gigliano G, Amadori S, Mandelli F. Flow cytometric measurement of microcytic and hyperchromic red cell populations in paediatric patients affected by hereditary spherocytosis (HS), Haematologica 1989; 74: 547-553.
- Kutter D, Mijanovic-Lazic D, Kremer A. Routinemässige Diagnose einer extravasalen Hämolyse durch die Erfassung von Hyperchromie und Sphärozytose. Berichte de ÖGKC 2002; 25: 5-10.
- Kutter D, Thoma J, Coulon N, Janecki J. La sphérocytose héréditaire – un diagnostic souvent méconnu. Bull Soc Lux Biol Clin 2002; 23: 75 – 83.
- Janecki J. A computerized graphic method for the extraction of a Gaussian part from the Gaussian-like distribution and its application in a medical laboratory Proceedings 7th National Conference on Application of Mathematics in Biology and Medicine. Zawoja (Poland) 2001.
- Kutter D, Mijanovic-Lasic D, Thoma J, Kutter P. Erythrocyte hyperchromia measured by flow cytometry: a marker for body iron overloa J Lab Med 2003; 27: 67-72.
- Kutter D, Mijanovic-Lasic D. Is there a link between spherocytosis and diabetes Bull Soc Lux Biol Clin 2003; 24: 22- 27.
- Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T. Penetrance of 845G®A(C282Y)HFE hereditary haemochromatosis mutations in the USA. Lancet 2002; 359: 211-218.
- Kang JO. Chronic iron overload and toxicity: clinical chemistry perspective, Clin Lab Sci 2001; 14: 209-219.
- Bottomley SS. Secondary iron overload disorders. Semin Hematol 1998; 35: 77-81.
- Weinberg ED. Development of clinical methods of iron deprivation for suppression of neoplastic and infectious disease. Cancer Invest 1999;17: 507-513.
- Weinberg ED. The role of iron in cancer. European Journal of Cancer Prevention 1996; 5: 19-36.
- Weinberg ED, Weinberg GA. The role of iron in infection. Current Opinion in Infectious Diseases 1995; 8: 164-168.
- Weinberg ED. Iron withholding: a defense against viral infections. BioMetals 1996; 9: 393-399.
- Deugnier Y, Moirand R. Surcharge martiale et santé publique. Bull Acad Natl Med 2000;184: 365-373.
- Sullivan JL. Iron and the sex difference in the heart disease risk, Lancet 1981;307:1293-1294.
- Tuomainen TP, Kontula K, Nyyssonen K, Lakka A,Helio T, Salonen TJ. Increased risk of acute myocardial.
- Salonen JT, Nyyssönen K, Korpela H, Tuomilehto J, Seppänen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation 1992; 86: 803-811.
- Passa P, Gourgon R, Cazor JL, Masquet C. Hémochromatose cardiaque. Résultats anatomiques, cliniques et hémodynamiques. Nouv, Press. Méd 1975; 4: 1017-1022.
- Meyers, DG. The iron hypothesis – does iron cause artherosclerosis Clin Cardiol 1996; 19: 925-929.
- Campell S, George DK, Robb SD. Spooner R, McDounagh TA, Dargie HJ, Mills PR. The prevalence of hamochromatosis gene mutations in the West of Scotland and their relation to ischemic heart disease. Blood 2003; 89: 1023-1026.
- Bryszewska M, Szosland K. Association between the glycation of erythrocyte membrane proteins and membrane fluidity. Clin Biochem 1988; 21: 49-51.
- Watala C. Hyperglycaemia alters the physico-chemical properties of proteins in erythrocyte membranes of diabetic patients. Int J Biochem 1992; 24: 1755-1761.
- Wazulikova I, Sikurova L, Carsky J, Strbova L, Krahulec B. Decreased fluidity of erythrocyte membranes in type 1 and type 2 diabetes. Gen Physiol Biophys 2000; 19: 381-392.
- Przybylska M, Bryszewska M, Chapman IV. Thermal properties and fluidity of human erythrocyte membranes in diabetes mellitus. Int J Radiat Biol 1993;63:419-424.
- Waugh RE, Agre P. Reduction of erythrocyte membrane viscoelastic coefficients reflects spectrin deficiencies in hereditary spherocytosis. J Clin Invest 1988; 81: 133-141.
- Yaouanq JM. Diabetes and haemochromatosis: current concepts, management and prevention. Diabetes Metab 1995; 21:319-329.
- Dubois-Laforgue D, Larger E, Timsit J. Le diabete sucré est-il une condition suffisante pour suspecter une hémochromatose Diabetes Metab 2000; 26: 318-321.
- Salonen JT, Tuomainen TP, Kontula K. Role of C282Y mutation in haemochromatosis gene in development of type 2 diabetes in healthy men: prospective cohort study. BMJ 2000; 320: 1706-1707.
- Conway AM, Vora AJ, Hinchliffe RF. The clinical relevance of an isolated increase in the number of circulating red blood cells. J Clin Pathol 2002; 55: 841-844.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC


