CITRATO DE SILDENAFIL EN MODELOS EXPERIMENTALES DE SINDROMES CORONARIOS AGUDOS
(especial para SIIC © Derechos reservados)
Coautor
Rakesh Kukreja*
PhD, Medical Collage of Virginia, Virginia Commonwealth University, Richmond, EE.UU.*
Recepción del artículo: 7 de abril, 2003
Aprobación: 0 de , 0000
 Conclusión breve
En modelos experimentales de angina inestable y de síndrome coronario agudo, el sildenafil no exacerba la isquemia.
Resumen
La investigación reciente prestó máxima atención a la seguridad y a las consecuencias cardiovasculares del citrato de sildenafil en el contexto de la enfermedad coronaria. En esta revisión, presentamos las evaluaciones realizadas en nuestros estudios de la acción del sildenafil en modelos experimentales de síndromes coronarios agudos. Las observaciones demuestran que el fármaco no exacerba la isquemia en perros con estenosis coronaria -modelo que simula la situación clínica de la "hibernación a corto plazo"- o con angina inestable. Más aún, brindamos evidencia del efecto protector del sildenafil en conejos y modelos murinos de infarto agudo de miocardio.
Palabras clave
Isquemia de miocardio, infarto de miocardio, flujo coronario, estenosis coronaria
Clasificación en siicsalud
Conclusión breve
En modelos experimentales de angina inestable y de síndrome coronario agudo, el sildenafil no exacerba la isquemia.
Resumen
La investigación reciente prestó máxima atención a la seguridad y a las consecuencias cardiovasculares del citrato de sildenafil en el contexto de la enfermedad coronaria. En esta revisión, presentamos las evaluaciones realizadas en nuestros estudios de la acción del sildenafil en modelos experimentales de síndromes coronarios agudos. Las observaciones demuestran que el fármaco no exacerba la isquemia en perros con estenosis coronaria -modelo que simula la situación clínica de la "hibernación a corto plazo"- o con angina inestable. Más aún, brindamos evidencia del efecto protector del sildenafil en conejos y modelos murinos de infarto agudo de miocardio.
Palabras clave
Isquemia de miocardio, infarto de miocardio, flujo coronario, estenosis coronaria
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/20128
Especialidades
 Principal: Farmacología,
Principal: Farmacología,
Relacionadas: Cardiología, Medicina Interna, Urología,
Enviar correspondencia a:
Karin Przyklenk, PhD. Department of Emergency Medicine, University of Massachussets Medical School, 55 Lake Avenue North, Worcester, MA. 01655 USA
SILDENAFIL CITRATE IN EXPERIMENTAL MODELS OF ACUTE CORONARY SYNDROMES
Abstract
Considerable recent interest has focused on the safety and cardiovascular consequences of sildenafil citrate in the setting of coronary artery disease. Our aim in this review is to summarize our experimental studies evaluating the effects of sildenafil in experimental models of acute ischemic syndromes. We report that sildenafil does not exacerbate ischemia in canine models of coronary artery stenosis mimicking clinical instances of \'short-term hibernation\' and unstable angina. Moreover, we provide evidence of sildenafil-induced cardioprotection in rabbit and murine models of acute myocardial infarction.
Key words
Isquemia de miocardio, infarto de miocardio, flujo coronario, estenosis coronaria
CITRATO DE SILDENAFIL EN MODELOS EXPERIMENTALES DE SINDROMES CORONARIOS AGUDOS
(especial para SIIC © Derechos reservados)
Artículo completo
Desde su comercialización en marzo de 1998, el citrato de sildenafil fue prescripto a aproximadamente 16 millones de hombres con disfunción eréctil, muchos de los cuales también presentaban enfermedad cardiovascular. Por este motivo -factores de riesgo compartidos y por la etiología subyacente común en los dos trastornos- se ha prestado atención considerable al uso del sildenafil en poblaciones específicas de enfermos.1-3 Aunque la evidencia hasta la fecha confirma el perfil benigno de la droga,2,3 las recomendaciones establecidas por la American Heart Association y el American College of Cardiology establecen la necesidad de su uso con cautela en pacientes con enfermedad coronaria e isquemia activa.1
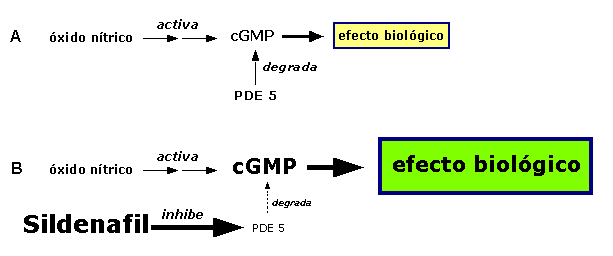
Figura 1. Mecanismo de acción del sildenafil. La señalización mediada por GMPc es: (A) activada por óxido nítrico y (B) amplificada por el sildenafil a través de la inhibición específica de la enzima PDE-5.cGMP, 3\'5\' monofosfato de guanosina cíclico. PDE-5, fosfodiesterasa 5.
Esta vigilancia tiene que ver con el mecanismo de acción del sildenafil (figura 1); el fármaco facilita la erección peneana al inducir relajación del músculo liso y vasodilatación en presencia de óxido nítrico (ON), fenómenos que obedecen a la inhibición selectiva de la enzima fosfodiesterasa-5 (PDE-5) que actúa sobre el 3\',5\'-monofosfato de guanosina cíclico (cGMP). La enzima se encuentra en concentraciones elevadas en el cuerpo cavernoso.1-3 Sin embargo, debido a que también se localiza en músculo liso vascular, el sildenafil puede originar simultáneamente vasodilatación de la circulación coronaria y sistémica. 1,2 Si bien una vasodilatación moderada puede ser beneficiosa al mejorar el flujo distal a un sector de estenosis coronaria4 y al desencadenar mecanismos cardioprotectores similares a los del preacondicionamiento,5 surge preocupación por la posibilidad de que el sildenafil comprometa la perfusión coronaria en el contexto de la isquemia de miocardio. Este efecto obedecería a la vasodilatación coronaria que inicia el mecanismo de robo coronario; también podría ser consecuencia secundaria de la hipotensión por vasodilatación sistémica.1-4 Los aspectos señalados motivaron numerosa investigación con la finalidad de comprender los efectos cardiovasculares del sildenafil en pacientes con enfermedad coronaria6-9 y, en menor medida, en modelos experimentales de síndromes coronarios agudos. En la revisión actual, describimos los estudios experimentales que realizamos al respecto en dos laboratorios separados. El objetivo fue evaluar el efecto del sildenafil en modelos caninos de estenosis coronaria (KP:4) y en el modelo de conejos con infarto agudo de miocardio (RCK:5). Sildenafil en modelos de estenosis coronaria
Hipoperfusión estable: un modelo de hibernación a corto plazo
En dos modelos a tórax abierto de perros anestesiados (estudio realizado en el Good Samaritan Hospital, Los Angeles CA)4 se determinaron las consecuencias del tratamiento con sildenafil sobre el flujo coronario. En el primer protocolo se colocó un micromanómetro constrictor alrededor de la coronaria descendente anterior izquierda (DA) hasta que el flujo coronario se redujo en aproximadamente un 50% respecto de los valores basales (figura 2A). La aplicación de estenosis en una arteria indemne se asocia on hipoperfusión estable (figura 2B) y representa el modelo clásico de "hibernación a corto plazo".10 Luego de una hora de pretratamiento libre de medicación, los animales fueron asignados a recibir dosis crecientes de sildenafil o placebo (sildenafil en dosis de 0.7 mg/kg disuelto en solución salina y administrado por vía intravenosa, iv).
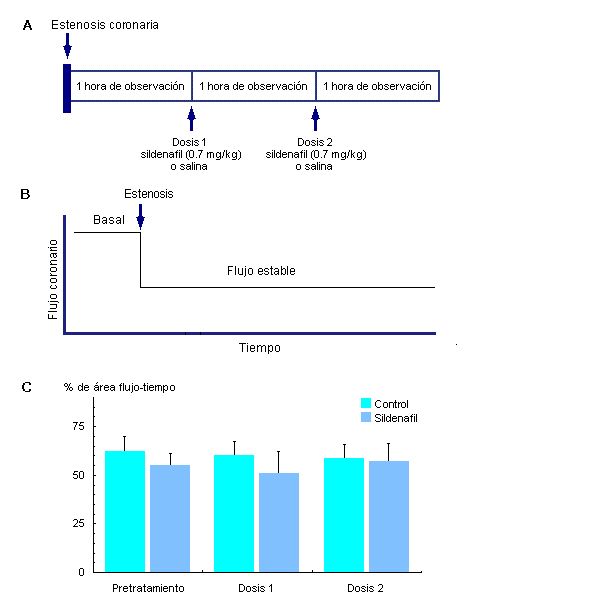
Figura 2. Modelo de estenosis coronaria que simula la situación clínica de la hibernación del miocardio a corto plazo. A, diseño del protocolo. B, aplicación de la estenosis en un segmento indemne de coronaria que se asocia con hipoperfusión estable. C, % de área flujo-tiempo (media ± DE) (índice de permeabilidad coronaria) medido durante 1 hora antes de la aleatorización y tratamiento (Pretratamiento), durante 1 hora después de la primer dosis de sildenafil o salina (Dosis 1) y durante 1 hora después de la segunda dosis de sildenafil o salina (Dosis 2). El panel C se reimprimió de la referencia #4, con autorización.
Los animales recibieron dos dosis de sildenafil o de solución salina una y dos horas después de la estenosis coronaria (figura 2A). La dosis se eligió de manera tal que se aproximara a la dosis de 50 mg de sildenafil que recibe un paciente de 70 kg. Debido a que la vida media del fármaco es de alrededor de 4 horas,1 la dosis acumulada de 1.4 mg/kg durante la hora final de observación se acerca a la dosis clínica de 100 mg.4 La perfusión coronaria durante los 60 minutos pretratamiento y durante la hora posterior a la primera y segunda dosis se cuantificó mediante el registro del perfil de flujo coronario. Luego, se normalizó el área flujo-tiempo para cada perro según su flujo basal por hora.4 La administración de sildenafil por vía iv generó una marcada pero transitoria (menos de 5 minutos) reducción de la presión arterial.4 Es de destacar que no hubo evidencia de que el sildenafil exacerbara la isquemia en este modelo: el área flujo-tiempo (índice de permeabilidad vascular) fue del 50% al 60% del basal a lo largo de todo el protocolo en todos los animales. No se registraron diferencias luego de la administración del sildenafil en comparación con el período inicial libre de tratamiento o con placebo (figura 2C).4 La observación de que el sildenafil no es dañino coincide con los resultados de otras investigaciones que utilizaron perros conscientes en los cuales el tratamiento con sildenafil se asoció con aumento de la perfusión coronaria en reposo y durante el ejercicio.11,12 Más aún, en hombres con enfermedad coronaria, el sildenafil no tuvo efecto coronario ni indujo vasodilatación moderada.7 Por lo tanto, bajo estas circunstancias, la evidencia no muestra agravamiento de la perfusión coronaria en relación con el tratamiento con sildenafil. Trombosis recurrente mediada por plaquetas: un modelo de angina inestable
El segundo protocolo experimental, realizado también en perros con tórax abierto, fue similar en diseño al descripto anteriormente pero, en este caso la DA inicialmente se comprimió con hemostats (que induce denudación endotelial y lesión arterial) antes de la colocación del micromanómetro constrictor (figuras 3A y 4).4

Figura 3. Modelo de lesión arterial más estenosis, situación que simula la angina inestable. A. Diseño del protocolo. B. La lesión arterial más la estenosis inicia el desarrollo de variaciones cíclicas en el flujo sanguíneo coronario como consecuencia de la formación repetida y desprendimiento de trombos ricos en plaquetas en el vaso dañado y estenosado. C. % de área flujo-tiempo (media ± EEM) (índice de permeabilidad coronaria) medido durante 1 hora antes de la aleatorización y tratamiento (Pretratamiento), durante 1 hora después de la primer dosis de sildenafil o salina (Dosis 1) y durante 1 hora después de la segunda dosis de sildenafil o salina (Dosis 2). * p < 0.05 versus control.** p < 0.05 versus pretratamiento.El panel C se reimprimió de la referencia #4, con autorización.

Figura 4. Corte histológico de una coronaria canina a nivel de la lesión y estenosis, con tinción de rojo de picrosirio y visto con iluminación en campo brillante. Con este método, la adventicia aparece de color rojo mientras que la capa media se tiñe de color amarillo-naranja. La capa media está disgregada (flechas llenas) y los remanentes de trombos ricos en plaquetas (flechas vacías) son visibles en la luz. Reimpreso de la referencia #14, con autorización.
Este escenario -de estenosis coronaria aplicada en un sitio de lesión vascular (el modelo Folts clásico) desencadena rápidamente (en menos de 5 minutos) variaciones cíclicas en el flujo coronario (CFVs) por la formación espontánea reiterada y desprendimiento de trombos ricos en plaquetas. El modelo simula muchos hallazgos fisiopatológicos de la isquemia breve y recurrente, característicos de la angina inestable (figuras 3B y 4).4,13-15 Al igual que en el protocolo anterior, los perros fueron sometidos a una hora inicial sin tratamiento luego de la lesión y estenosis arterial; posteriormente cada animal fue asignado a sildenafil (0.7 mg/kg por vía iv una y dos horas posestenosis) o salina (figura 3A). La permeabilidad vascular fue, nuevamente, valorada por la medición del área de flujo coronario durante cada período.4 Como era de esperar, todos los perros tuvieron CFVs como consecuencia de la lesión y estenosis vascular; el área flujo-tiempo durante la hora pretratamiento fue de alrededor 40% en ambas poblaciones de animales. El grupo que recibió placebo presentó aumento temporario moderado en el área flujo-tiempo (a aproximadamente 60% del basal durante la última hora de observación; figura 3C) 4, un efecto desencadenado por la liberación de adenosina a partir del miocardio con isquemia y reperfusión durante los episodios precoces de oclusión trombótica y reperfusión. Como consecuencia, se inhibe parcialmente la agregación plaquetaria mediante la estimulación de los receptores de adenosina A2 en la superficie plaquetaria.14-16 El sildenafil no exacerbó la isquemia en el contexto de la trombosis recurrente: el área de flujo-tiempo se mantuvo prácticamente sin cambios, al 35% aproximadamente, luego de las dos administraciones del fármaco. Resultó llamativa la comprobación de que los animales tratados con sildenafil no presentaron el aumento transitorio y moderado en la permeabilidad vascular mediada por los receptores de adenosina A2 tal como se observó en controles (figura 3C).4 Sildenafil, adenosina y agregación plaquetaria
Fue evidente que la perfusión coronaria no empeoró en relación con el tratamiento con sildenafil. Sin embargo, el hallazgo sutil e inesperado de la aparente falta de efecto inhibitorio plaquetario mediado por la estimulación de receptores de adenosina en el grupo de animales tratados motivó estudios adicionales en relación con la acción del sildenafil sobre la reactividad de las plaquetas.4 Las plaquetas tienen altas concentraciones de PDE-5 y, de hecho, los inhibidores de la PDE-5 han sido propuestos como una nueva modalidad de terapia antiplaquetaria.17,18 No obstante, los resultados observados con el sildenafil han sido mixtos: algunas investigaciones revelaron una inhibición moderada de la activación y agregación de plaquetas7,9 mientras que otras no encontraron efecto alguno.3,17 Nuestras observaciones in vivo avalan la idea de que el sildenafil no atenúa la agregación y activación plaquetaria en coronarias estenosadas y lesionadas. Los hallazgos se confirmaron in vitro mediante agregometría sanguínea: la agregación de plaquetas no se afectó por el agregado exógeno de sildenafil como así tampoco en muestras de sangre obtenidas de perros tratados con sildenafil.4 La evidencia in vivo en modelos caninos también sugiere que el sildenafil puede hacer que las plaquetas pierdan la capacidad de responder a los efectos inhibitorios mediados por la estimulación de los receptores de adenosina A2. Este concepto se confirmó in vitro al comprobarse que el agregado exógeno de un agonista de tales receptores -CGS 21680- desencadenaba una reducción del 25% de la agregación plaquetaria en muestras control pero no en muestras de sangre con sildenafil o en muestras de animales pretratados con sildenafil (figura 5).4

Figura 5. Parte superior: Registros originales de la agregación plaquetaria in vitro (aumento de la impedancia) en muestras de sangre. En condiciones basales, la agregación plaquetaria es atenuada por el agregado exógeno del agonista de los receptores A2 de adenosina -CGS 21680- (panel derecho) versus el agregado de un volumen comparable de vehículo (salina: panel izquierdo). Las flechas indican el agregado de colágeno como estímulo para el inicio de la agregación de plaquetas. Barra = 2 minutos. Parte inferior: % de cambio en la agregación plaquetaria in vitro con el agregado exógeno del agonista de los receptores A2 de adenosina -CGS 21680- en muestras de sangre de perros control y tratados con sildenafil. Los datos se obtuvieron antes de la aleatorización y tratamiento (Pretratamiento) y después de la segunda dosis del fármaco o salina (dosis 2) y reflejan el % de diferencia en la impedancia en muestras pareadas con el agregado de CGS versus vehículo (salina). El panel inferior adaptado de la referencia #4, con autorización.
¿Cómo podría explicarse este efecto inesperado y confuso del fármaco en la inhibición de la agregación plaquetaria mediada por A2 Aún se desconoce el mecanismo por el cual el sildenafil (que presuntamente aumenta el contenido de GMPc por la inhibición de la PDE-5, figura 1) podría bloquear la inhibición de la agregación plaquetaria mediada por los receptores A2 (que se logra con la estimulación de la adenilato ciclasa y con aumento de la expresión del monofosfato de adenosina cíclico, AMPc).18 Sin embargo, hay evidencia de una interacción o "comunicación" entre el GMPc y el AMPc: el aumento en el nivel de GMPc que se observa con sildenafil induce un incremento (más que descenso) en la cantidad de AMPc.20-22 Empero, cada vez hay más evidencia de que el aumento del GMPc puede ejercer un efecto dual bifásico sobre las plaquetas. Específicamente, el efecto inhibitorio plaquetario bien descripto que se observa con el aumento del GMPc va precedido de una estimulación inicial y transitoria de la agregación de plaquetas.23 Este fenómeno hace surgir la posibilidad alternativa de que el sildenafil per se no torna a las plaquetas refractarias a la estimulación de los receptores A2 de adenosina. En cambio, la falta de mejoría en la permeabilidad coronaria en los perros tratados con sildenafil podría deberse a una moderada estimulación de la agregación de plaquetas, mediada por el GMPc, que compensa el efecto inhibitorio de la estimulación a través de los receptores A2. Sin embargo, esta explicación es especulativa y se requiere mayor investigación al respecto. Sildenafil en modelos de infarto agudo de miocardio
La hipótesis
Durante los últimos 15 años, el interés se centralizó esencialmente en los efectos cardioprotectores marcados del preacondicionamiento isquémico (PC), durante el cual breves episodios de isquemia hacen que el miocardio se torne más resistente a una agresión isquémica posterior sostenida.24 Si bien aún no se comprenden por completo los mecanismos celulares responsables en este fenómeno, se ha avanzado notoriamente en las vías de señalización intracelular que contribuyen con esta cardioprotección paradójica. A su vez, se han identificado desencadenantes farmacológicos (adenosina, bradiquinina) capaces de iniciar y simular los beneficios del PC inducido por la isquemia convencional.25-29 Sin embargo, el concepto de isquemia y de PC farmacológico aún no se refleja con peso en la clínica, probablemente como consecuencia de la falta de drogas eficaces (con aprobación de la Food and Drugs Administration) con perfil farmacológico que simule el efecto del PC. A pesar de la prudencia recomendada cuando se administra sildenafil a personas con enfermedad coronaria1 aún cabe la pregunta de si el fármaco podría actuar como un agente protector al inducir un efecto símil PC. La racionalidad de esta teoría, propuesta por RCK5 y mostrada esquemáticamente en la figura 6 consiste en que la acción vasodilatadora del sildenafil podría aumentar la liberación de mediadores endógenos de PC, tales como adenosina o bradiquinina a partir de las células endoteliales y desencadenar así una cascada de señalización (a través de la acción de quinasas) que culmina con la fosforilación de la sintetasa de ON y con la liberación de ON. La formación de este último mediador podría, en teoría, activar la guanilato ciclasa con mayor producción de GMPc, activación de la proteinquinasa C y, finalmente, apertura de los canales mitocondriales de potasio dependientes de ATP (KATP),30 involucrados como un posible efector final del PC isquémico convencional.31

Figura 6.: Ilustración esquemática de los mecanismos celulares propuestos por los cuales el sildenafil podría asociarse con cardioprotección. cGMP, 3\'5\' monofosfato de guanosina cíclico. eNOS, sintetasa de óxido nítrico endotelial. GTP, trifosfato de guanosina. 5-HD. 5-hidroxidecanoato. Canal KATP, canal de potasio sensible al ATP; NO, óxido nítrico. PDE-5, fosfodiesterasa 5.
Sildenafil y cardioprotección
La hipótesis se evaluó in vivo en el modelo de infarto de miocardio en conejos.5 En forma específica, los animales fueron tratados con sildenafil (0.7 mg/kg) administrado por vía oral o iv. Entre 30 y 60 minutos después del tratamiento (fase aguda) o a las 24 horas (fase tardía), los animales fueron sometidos a 30 minutos de oclusión coronaria sostenida seguida de 3 horas de reperfusión. El tamaño del infarto -principal variable de evaluación- se determinó mediante marcación con trifeniltetrazolio; el área de necrosis se expresó como el porcentaje del territorio de la coronaria ocluida (área en riesgo de infarto por la marcación con infusión de colorante).5 Como era de esperar, los conejos tratados con sildenafil mostraron hipotensión: 5 la administración iv se asoció con rápido pero breve (de 2 a 5 minutos) descenso de la presión arterial media, sistólica y diastólica (figura 7) similar a los resultados que se obtuvieron en el modelo canino.4 En cambio, el tratamiento por vía oral se acompañó de una respuesta hemodinámica gradual, moderada y más prolongada. De manera llamativa, el sildenafil produjo un efecto cardioprotector considerable.5

Figura 7. Presión arterial sistólica, diastólica y media (PAS, PAD y PAM, respectivamente) medidas antes (tiempo 0), 2 minutos y 5 minutos después de la administración de sildenafil por vía intravenosa en el modelo de conejos. *p < 0.05 versus tiempo 0.Reimpreso de la referencia #5, con autorización.
Durante la fase de tratamiento iv, el tamaño del área de infarto fue de 34 ± 2% de la región de riesgo en los animales tratados con salina versus sólo 11 ± 1% en la fase aguda del tratamiento con sildenafil y 20 ± 2% en la fase tardía de la terapia (figuras 8A y 9).

Figura 8. Area media de infarto expresada como % de la región de riesgo luego de la administración de sildenafil por vía intravenosa (A) y por vía oral (B). * p < 0.05 >versus control.5-HD, 5-hidroxidecanoato.Reimpreso de la referencia #5, con autorización.
De hecho, se constató reducción en el tamaño del infarto de 68% a 41%, respectivamente. La administración oral del sildenafil se asoció con los mismos efectos beneficiosos (figura 8B).5 Aún no se sabe si la reducción más marcada en el tamaño del infarto que se observa en la fase aguda del tratamiento con sildenafil versus la fase tardía representa declinación gradual del efecto protector sostenido o si la protección inducida por sildenafil es bifásica (análoga a la primera ventana clásica y a la segunda ventana tardía del PC isquémico). Más aún, no se comprende si en la protección de las fases aguda y tardía actúan mecanismos distintos. Ambos fenómenos indudablemente requieren mayor investigación. Primeras observaciones en relación con los mecanismos responsables
Con la finalidad de entender los mecanismos por los cuales el sildenafil induce limitación del tamaño de la zona infartada y, en particular, para explorar la posible contribución de los canales mitocondriales de KATP, un grupo adicional de conejos tratados con sildenafil recibió el antagonista de dichos canales, 5-hidroxidecanoato (5-HD, 5 mg/kg por vía iv) 10 minutos antes de la isquemia y reperfusión. Fue interresante comprobar que el efecto limitante del sildenafil sobre la zona de infarto en la fase aguda y tardía quedó abolido en animales que recibieron 5-HD: el tamaño del infarto fue de alrededor del 36% de la región de riesgo en los animales tratados con sildenafil y 5-HD, en comparación con los valores observados en los animales del grupo control (figuras 8A y 9).5

Figura 9. Fotografías originales de corazones de conejos que demuestran la reducción del tamaño del infarto con sildenafil y el bloqueo del efecto protector con 5-hidroxidecanoato (5-HD). Los corazones se perfundieron con azul de Evans para delinear la región de riesgo, se cortaron en forma transversal y se tiñeron con cloruro de trifeniltetrazolio para marcar la zona de necrosis. Las áreas azuladas representan tejido normalmente perfundido, las regiones rojizas indican miocardio viable mientras que la zona infartada permanece sin tinción por lo que se la observa gris pálida. Reimpreso de la referencia #5, con autorización.
¿Como puede extrapolarse dicho fenómeno -aparentemente un componente importante de la cardioprotección mediada por sildenafil- a la disminución del tamaño del infarto Es bien sabido que las mitocondrias intervienen de manera esencial en la sobrevida de las células por la síntesis de ATP y por mantener la homeostasis del calcio (Ca2+). La apertura de los canales mitocondriales KATP compensa en forma parcial el potencial de membrana y permite la salida adicional de protones con la finalidad de que se genere un gradiente electromecánico para la síntesis de ATP y para el transporte de Ca2+.31 La protección aguda inducida por sildenafil puede estar mediada por la apertura de los canales mitocondriales de KATP ya sea en forma directa o mediante diversas vías de señalización tales como activación de la proteinquinasa C y de quinasas activadas por mitógenos. En cambio, en la fase tardía podría intervenir una cascada de señalización que culmina con la expresión de la sintetasa inducible de ON, la producción de ON y la apertura de los canales mitocondriales de KATP en la forma que se describió previamente.27.28,32-35 Aunque por ahora no hay evidencia que avale esta teoría, estudios finalizados recientemente en el modelo murino de isquemia y reperfusión confirmaron las observaciones obtenidas en conejos en relación con el efecto cardioprotector del sildenafil. Más notablemente, se reconoció que la mayor expresión de la sintetasa de ON se relaciona con la disminución del tamaño del área de infarto inducida por el sildenafil.36Resumen y perspectivas futurasAunque las recomendaciones de la American Heart Association y del American College of Cardiology son, sin duda, prudentes, los hallazgos comentados no avalan que el sildenafil se asocie con consecuencias cardiológicas deletéreas en modelos animales de síndromes coronarios agudos. El sildenafil no exacerbó la isquemia en modelos caninos de hipoperfusión coronaria estable o en el modelo de trombosis recurrente mediada por plaquetas.4,5 Sin embargo, uno de los puntos aún oscuros en este perfil aparentemente benigno del sildenafil (el efecto moderado en la inhibición de la agregación de plaquetas mediada por receptores A2 de adenosina,4) y su posible importancia clínica merecen mayor investigación. En segundo lugar, el sildenafil no empeora la injuria por isquemia en conejos y modelos murinos de infarto agudo de miocardio. Por el contrario, en este contexto, el sildenafil se asocia con un marcado efecto cardioprotector a través de la apertura de los canales mitocondriales de KATP y de la mayor síntesis de ON, fenómenos celulares importantes involucrados en dicho fenómeno.5,36 Sin embargo, parece prematuro concluir, en función de los hallazgos observados a la fecha, que el efecto cardioprotector del sildenafil en modelos experimentales pueda trasladarse a pacientes con enfermedad isquémica. Sin embargo, estas observaciones abren nuevas perspectivas acerca del potencial efecto beneficioso del sildenafil, como un cardioprotector que induce un fenómeno similar al del PC.
Bibliografía del artículo
- Cheitlin MD, Hutter AM, Brindis RG et al. Use of sildenafil (Viagra) in patients with cardiovascular disease. J Am Coll Cardiol 1999;33:273-282.
- Kloner RA. Cardiovascular risk and sildenafil.. Am J Cardiol 2000;86 (Suppl F): 57F-61F.
- Gillies HC, Roblin D, Jackson G. Coronary and systemic hemodynamic effects of sildenafil citrate: from basic science to clinical studies in patients with cardiovascular disease. Int J Cardiol 2002;86:131-141.
- Przyklenk K, Kloner RA. Sildenafil citrate (Viagra) does not exacerbate myocardial ischemia in canine models of coronary artery stenosis. J Am Coll Cardiol 2001;37:286-292.
- Ockaili R, Salloum F, Hawkins J, Kukreja RC. Sildenafil (Viagra) indices powerful cardioprotective effect via opening of mitochondrial KATP channels in rabbits. Am J Physiol 2002;283:H1263-H1269.
- Hermann HC, Chang G, Klugherz BD, Mahoney PD. Hemodynamic effects of sildenafil in men with severe coronary artery disease. New Engl J Med 2000;342:1622-1626.
- Halcox JPJ, Nour KRA, Zalos G et al. The effect of sildenafil on human vascular function, platelet activation and myocardial ischemia. J Am Coll Cardiol 2002;40:1232-1240.
- Bocchi EA, Guimarães G, Mocelin A, Bacal F, Bellotti G, Ramires JF. Sildenafil effects on exercise, neurohumoral activation and erectile dysfunction in congestive heart failure: a double-blind, placebo-controlled, randomized study followed by a prospective treatment for erectile dysfunction. Circulation 2002;106:1097-1103.
- Arruda-Olson AM, Mahoney DW, Nehra A, Leckel M, Pellikka PA. Cardiovascular effects of sildenafil during exercise in men with known or probable coronary artery disease. JAMA 2002;287:719-725.
- Heusch G. Hibernating myocardium. Physiol Rev 1998;78:1055-1085.
- Chen Y, Du R, Traverse JH, Bache RJ. Effect of sildenafil on coronary active and reactive hyperemia. Am J Physiol 2000;279:H2319-H2325.
- Traverse JH, Chen YJ, Du R, Bache RJ. Cyclic nucleotide phosphodiesterase type 5 activity limits blood flow to hypoperfused myocardium during exercise. Circulation 2000;102:2997-3002.
- Folts JD. An in vivo model of experimental arterial stenosis, intimal damage and periodic thrombosis. Circulation 1991;83 (Suppl IV):IV3-IV14.
- Hata K, Whittaker P, Kloner RA, Przyklenk K. Brief antecedent ischemia attenuates platelet-mediated thrombosis in damaged and stenotic canine coronary arteries: role of adenosine. Circulation 1998;97:692-702.
- Ikeda H, Koga Y, Kuwano K et al. Cyclic flow variations in conscious dog model of coronary artery stenosis and endothelial injury correlate with acute ischemic heart disease syndromes in humans. J Am Coll Cardiol 1993;21:1008-1017.
- Kitakaze M, Hori M, Sato H, et al. Endogenous adenosine inhibits platelet aggregation during myocardial ischemia in dogs. Circ Res 1991;69:1402-1409.
- Wallis RM, Corbin JD, Francis SH, Ellis P. Tissue distribution of phosphodiesterase families and the effects of sildenafil on tissue cyclic nucleotides, platelet function and the contractile responses of trabeculae carnae and aortic rings in vitro. Am J Cardiol 1993;83 (Suppl 5A):3C-12C.
- Haslam RJ, Dickinson NT, Jang EK. Cyclic nucleotides and phosphodiesterases in platelets. Thromb Haemostasis 1999;82:4123-4132.
- Berkels R, Klotz T, Engelmann U, Klaus W. Modulation of human platelet aggregation by the phosphodiesterase type 5 inhibitor sildenafil. J Cardiovasc Pharmacol 2001;37:413-421.
- Stief CG, Uckert S, Becker AJ et al. Effect of sildenafil on cAMP and cGMP levels in isolated human cavernousum and cardiac tissue. Urology 2000;55:146-150.
- Schalcher C, Schad K, Brunner-La Rocca HP et al. Interaction of sildenafil with cAMP-mediated vasodilation in vivo. Hypertension 2002;40:763-767.
- Sugiyama A, Takeuchi N, Saegusa Y, Sugita M, Hashimoto K. Molecular mechanisms of cardiostimulatory effects of sildenafil. Jpn J Pharmacol 2002;88:362-364.
- Li Z, Xi X, Gu M et al. A stimulatory role for cGMP-dependent protein kinase in platelet activation. Cell 2003;112:77-86.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986;74:1124-1136.
- Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM. Acetylcholine, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial KATP channels. Circ Res 2001;89:273-278.
- Janin Y, Qian YZ, Hoag J, Elliott GT, Kukreja RC. Pharmacologic preconditioning with monophosphoryl lipid A is abolished by 5-hydroxydecanoate, a specific inhibitor of the KATP channel. J Cardiovasc Pharmacol 32;1998:337-342.
- Tajero-Taldo MI, Gursoy E, Kukreja RC. a-adrenergic receptor stimulation produces late preconditioning through inducible nitric oxide synthase in mouse heart. J Mol Cell Cardiol 2002;34:185-195.
- Zhao TC, Kukreja RC. Late preconditioning elicited by activation of adenosine A3 receptor in heart: role of NF- kB, iNOS and mitochondrial KATP channel. J Mol Cell Cardiol 2002;34:263-277.
- Kostiprapa C, Ockaili R, Kukreja RC. Bradykinin B2 receptor is involved in the late phase of preconditioning in rabbit heart. J Mol Cell Cardiol 2001;33:1355-1362.
- Han J, Kim N, Kim E, Ko WK, Earm YE. Modulation of ATP-sensitive potassium channels by cGMP-dependent protein kinase in rabbit ventricular myocytes. J Biol Chem 2110;726:22140-22147.
- Scewczyk A. The ATP-regulated K+ channel in mitochondria: five years after its discovery. Acta Biochim Pol 1996;43:713-719.
- Xi L, Jarrett NC, Hess ML, Kukreja RC. Essential role of inducible nitric oxide synthase in monophosphoryl lipid A-induced late cardioprotection: evidence from pharmacological inhibition and gene knockout mice. Circulation 1999;99:2157-2163.
- Xi L, Salloum F, Tekin D, Jarrett NC, Kukreja RC. Glycolipid RC-552 induces delayed preconditioning-like effect via iNOS-dependent pathway in mice. Am J Physiol 1999;277:H2418-H2424.
- Zhao TC, Taher MM, Valerie KC, Kukreja RC. p38 Triggers late preconditioning elicited by anisomycin in heart: involvement of NF-B and iNOS. Circ Res 2001;89:915-922.
- Zhao TC, Xi L., Chelliah J., Levasseur JE, Kukreja RC. Inducible nitric oxide synthase mediates delayed protection induced by activation of adenosine A1 receptors: evidence from gene knockout mice. Circulation 2000;102:902-907.
- Salloum F, Yin C, Xi L, Kukreja RC. Sildenafil induces delayed preconditioning through inducible nitric oxide synthase-dependent pathway in mouse heart. Circ Res 2003; 92: in press, April.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC


