POLIMORFISMO DE REPETIÇÃO NA REGIÃO TATA-BOX DO GENE UGT1A1 E SUA RELAÇÃO COM DIFERENTES SITUAÇÕES CLINICAS RELACIONADAS COM HIPERBILIRRUBINEMIA
(especial para SIIC © Derechos reservados)
Coautores
Emilia Vieira.* Ana Margarida Alexandrino.** Ana Sarmento.*** Rosa Branca.**** Ermelinda Santos-Silva y José Barbot.***** Rosário dos Santos******
Licenciatura em Análises Clínicas e Estudante de Mestrado da Universidade de Aveiro. Unidade de Genética Molecular do Instituto de Genética Médica Dr. Jacinto de Magalhães*
Licenciatura em Medicina. Unidade de Neonatologia da Maternidade Júlio Dinis**
Licenciatura em Medicina.Serviço de Pediatria do Hospital de Vila Real.***
Licenciatura em Medicina..Serviço de Hematologia do Hospital de Crianças Maria Pia.****
Licenciatura em Medicina. Serviço de Pediatria do y Serviço de Hematologia Hospital de Crianças Maria Pia*****
Mestrado em Genética Humana.Unidade de Genética Molecular do Instituto de Genética Médica Dr. Jacinto de Magalhães******
Recepción del artículo: 15 de marzo, 2004
Aprobación: 0 de , 0000
 Conclusión breve
A pesquisa da inserção alelo TA, efectuada por técnicas de biologia molecular simples, permite-nos estabelecer o diagnóstico de Síndroma de Gilbert assim como fazer o estudo da sua influência na gravidade e morbilidade da hiperbilirrubinémia de outras situações clínicas com ela relacionadas.
Resumen
Durante muitos anos o diagnóstico de síndroma de Gilbert (SG) foi de exclusão, dada a dificuldade no estudo bioquímico do enzima uridina difosfato-glucuronosil transferase-1 (UGT1A1). A localização do seu gene e posteriormente a identificação de mutações relacionadas com SG, veio alterar de forma significativa este panorama, principalmente quando em 1995 foi identificada uma inserção de dois nucleotídeos (TA) na região TA(6)TAA do seu promotor, que veio a revelar-se como a principal causa de SG em todas as populações estudadas. No ano 2000 iniciamos a pesquisa desta inserção. Paralelamente ao seu estudo em doentes com diagnóstico clínico de SG, procuramos avaliar a sua influência no quadro clínico de outras situações também relacionadas com hiperbilirrubinémia, nomeadamente na icterícia neonatal (IN) e na doença hemolítica crónica (DHC). A pesquisa da inserção TA foi efectuada em 354 amostras; 91 de doentes com diagnóstico clínico de SG, 45 de doentes com DHC, 18 de doentes com défice de glicose-6-fosfato desidrogenase com referência a IN (incluindo 5 com DHC), 100 de recém-nascidos (RN) com IN (23 dos quais com IN prolongada) e 100 RN com ausência de IN documentada. A pesquisa da inserção TA, efectuada por técnicas de biologia molecular simples, permite-nos estabelecer o diagnóstico de SG assim como fazer o estudo da sua influência na gravidade e morbilidade da hiperbilirrubinémia de outras situações clínicas com ela relacionadas. A análise dos nossos resultados sugere-nos que a presença do alelo [TA]7 é a principal responsável por SG na nossa população, está associada ao desenvolvimento de IN prolongada e, nos doentes com DHC, aumenta os níveis de bilirrubina e consequentemente o risco de desenvolvimento de litíase vesicular.
Palabras clave
Hiperbilirrubinémia, síndroma de Gilbert, icterícia neonatal, doença hemolítica crónica, UGT1A1
Clasificación en siicsalud
Conclusión breve
A pesquisa da inserção alelo TA, efectuada por técnicas de biologia molecular simples, permite-nos estabelecer o diagnóstico de Síndroma de Gilbert assim como fazer o estudo da sua influência na gravidade e morbilidade da hiperbilirrubinémia de outras situações clínicas com ela relacionadas.
Resumen
Durante muitos anos o diagnóstico de síndroma de Gilbert (SG) foi de exclusão, dada a dificuldade no estudo bioquímico do enzima uridina difosfato-glucuronosil transferase-1 (UGT1A1). A localização do seu gene e posteriormente a identificação de mutações relacionadas com SG, veio alterar de forma significativa este panorama, principalmente quando em 1995 foi identificada uma inserção de dois nucleotídeos (TA) na região TA(6)TAA do seu promotor, que veio a revelar-se como a principal causa de SG em todas as populações estudadas. No ano 2000 iniciamos a pesquisa desta inserção. Paralelamente ao seu estudo em doentes com diagnóstico clínico de SG, procuramos avaliar a sua influência no quadro clínico de outras situações também relacionadas com hiperbilirrubinémia, nomeadamente na icterícia neonatal (IN) e na doença hemolítica crónica (DHC). A pesquisa da inserção TA foi efectuada em 354 amostras; 91 de doentes com diagnóstico clínico de SG, 45 de doentes com DHC, 18 de doentes com défice de glicose-6-fosfato desidrogenase com referência a IN (incluindo 5 com DHC), 100 de recém-nascidos (RN) com IN (23 dos quais com IN prolongada) e 100 RN com ausência de IN documentada. A pesquisa da inserção TA, efectuada por técnicas de biologia molecular simples, permite-nos estabelecer o diagnóstico de SG assim como fazer o estudo da sua influência na gravidade e morbilidade da hiperbilirrubinémia de outras situações clínicas com ela relacionadas. A análise dos nossos resultados sugere-nos que a presença do alelo [TA]7 é a principal responsável por SG na nossa população, está associada ao desenvolvimento de IN prolongada e, nos doentes com DHC, aumenta os níveis de bilirrubina e consequentemente o risco de desenvolvimento de litíase vesicular.
Palabras clave
Hiperbilirrubinémia, síndroma de Gilbert, icterícia neonatal, doença hemolítica crónica, UGT1A1
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/67367
Especialidades
 Principal: Hematología, Pediatría,
Principal: Hematología, Pediatría,
Relacionadas: Bioquímica, Diagnóstico por Laboratorio, Genética Humana,
Enviar correspondencia a:
Dr. Elísio Costa. Rua da Boavista, 827, 4050-111 Porto, Portugal Sousa Costa, Elísio Manuel
TATA-BOX POLYMORPHISM IN UGT1A1 GENE IN PATIENTS WITH HYPERBILIRUBINEMIA ASSOCIATED CONDITIONS
Abstract
For many years Gilbert's Syndrome (GS) was a diagnosis made by exclusion. This had to do with the unavailability of biochemical methods for the study of the implied enzyme bilirubin UDP-glucuronosyl transferase (UGT1A1). Gene mapping with subsequent knowledge of GS associated mutations significantly altered this diagnostic approach. In 1995, a two nucleotide (TA) insertion in the TA(6)TAA promoter region of the UGT1A1 gene was identified, which was found to be the main cause of GS in all studied populations. By the year 2000 we started to search for this insertion, both in patients with the clinical diagnosis of GS and in patients with other hyperbilirubinemia associated conditions, namely Neonatal Jaundice (NJ) and Chronic Hemolytic Diseases (CHD). We tested 354 different blood samples for the presence of the TA insertion mutation: 91 from patients with clinical diagnosis of GS; 45 from patients with CHD; 18 with Glucose-6-phosphate dehydrogenase deficiency with history of NJ (five of them with documented CHD); 100 from newborns with NJ (including 23 with prolonged NJ); and 100 healthy newborns without documented NJ. Study of the TA insertion mutation, made by simple molecular biology techniques, allows us to make the diagnosis of GS as well as to study the influence of its presence in the clinical and analytical manifestations of hyperbilirubinemia in different clinical settings. Our results suggest that the presence of the (TA)7 allele is the main cause of GS in our population. Furthermore, it may be related to prolonged NJ and, in patients with CHD, to higher serum bilirubin levels and higher risk of colelithiasis.
Key words
Gilbert's syndrome, UGT1A1, hyperbilirubinemia, neonatal jaundice, chronic hemolytic disease
POLIMORFISMO DE REPETIÇÃO NA REGIÃO TATA-BOX DO GENE UGT1A1 E SUA RELAÇÃO COM DIFERENTES SITUAÇÕES CLINICAS RELACIONADAS COM HIPERBILIRRUBINEMIA
(especial para SIIC © Derechos reservados)
Artículo completo
Introdução
A bilirrubina é um tóxico natural produzido durante o metabolismo do grupo heme da hemoglobina ou de outras proteínas relacionadas com o grupo heme. Os seus níveis séricos aumentam quando existe uma necessidade aumentada de metabolizar grupos heme ou então, quando a sua excreção está de alguma forma afectada. Níveis muito elevados podem originar neurotoxidade, de moderada a letal.1-3
O enzima UDP-glucoronosiltransferase-1 (UGT1A1) é responsável pela glucuronidação da bilirrubina. Défices ligeiros deste enzima estão associados com síndroma de Gilbert (SG) e défices graves com o síndroma de Crigler-Najjar (SCN).4,5
Dada a dificuldade no estudo bioquímico do enzima UGT1A1, o diagnóstico de SG foi durante muitos anos de exclusão. A localização do seu gene e posteriormente a identificação de mutações relacionadas com o SG, veio alterar de forma significativa este panorama. A primeira mutação no gene do UGT1A1 foi descrita em 1992.6 Tratava-se de uma mutação nonsense encontrada em homozigotia num doente com SCN. No entanto, só em 1995 é que foram descritas as primeiras mutações neste gene relacionadas com o SG. Uma em particular, uma inserção de 2 nucleotídeos (TA) na região promotora do gene,7 tem vindo a revelar-se como a principal causa de SG nas populações ocidentais estudadas.8,-12 Esta inserção na região A(TA)nTAA do promotor do gene do UGT1A1 dificulta a ligação do factor transcripcional IID, um importante factor no início da transcrição. Isto condiciona uma diminuição na transcrição do gene, com consequente diminuição da actividade do enzima UGT1A1 e aumento dos níveis de bilirrubina, tanto em indivíduos normais como em portadores de outras patologias que condicionem hiperbilirrubinémia.13-21 A frequência estimada desta mutação é de 38.7% na população Europeia, de 16% na população de origem Asiática e de 42.6% na população de origem Africana. Beutler et al. previu uma frequência de homozigotia na população caucasiana de 15%.9
No ano 2000 iniciamos a pesquisa desta inserção (Figura 1). Paralelamente ao seu estudo em doentes com diagnóstico clínico de SG, procuramos avaliar a sua influência no quadro clínico de outras situações também relacionadas com hiperbilirrubinémia, nomeadamente na icterícia neonatal (IN) e na doença hemolítica crónica (DHC). Neste trabalho é feito um levantamento dos resultados obtidos e avaliada a sua importância face aos diferentes contextos clínicos.

Figura 1. Cromatogramas do tamanho do fragmento obtido no programa ABI GeneScan, correspondentes aos diferentes genótipos encontrados.
Síndroma de Gilbert
Até ao momento, estudamos 91 doentes com diagnóstico clínico de SG.21 A sua maioria, 89.01% (81/91), revelou-se homozigótica para a inserção TA ([TA]7/[TA]7). Dos restantes 10 doente, um revelou-se heterozigoto composto para uma e duas inserções ([TA]7/[TA]8) e nove revelaram-se heterozigóticos [(TA)6/(TA)7]. Nestes últimos, os estudos adicionais efectuados não permitiram identificar, até ao momento, alterações adicionais no gene UGT1A1. Apenas num destes casos foi encontrado heterozigotia para um polimorfismo (–1126 C>T) na região promotora do gene (Figura 2). O estudo populacional efectuado revelou que este polimorfismo estava presente em 4.39 % dos 114 cromossomas estudados. A frequência muito elevada de homozigotia para o alelo (TA)7 na nossa população de doentes com SG é sobreponível ao descrito na literatura para outras populações caucasianas. Isto tem condicionado que a pesquisa deste alelo, efectuada por técnicas de biologia molecular simples, se tenha transformado num exame de extrema utilidade na confirmação do diagnóstico de SG, particularmente das suas formas mais ligeiras.
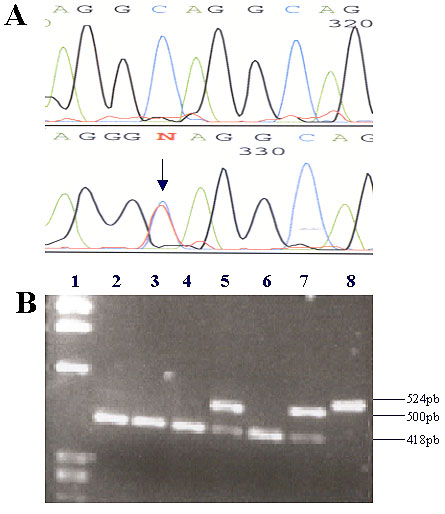
Figura 2. A: Extracto do electroforograma da sequenciação da região promotora do gene UGT1A1, onde se observa a substituição de uma citosina por uma timina (–1126 C>T). B: esta alteração elimina um local reconhecido pela enzima Cac8 I. 1- marcador de peso molecular. 2, 3 ,4 e 6- ausência do polimorfismo (418 pb + 82 pb + 24 pb). 5 e 7 – heterozigotia para o polimorfismo (500 pb + 24 pb). 8 – fragmento não digerido (524 pb).
Neste grupo de doentes encontramos uma proporção significativamente maior de indivíduos do sexo masculino (71.74%). Este facto está possivelmente relacionado com o facto dos indivíduos do sexo masculino homozigóticos para o alelo (TA)7 apresentarem valores de bilirrubina total mais elevados que os indivíduos do sexo feminino.
A presença do alelo (TA)8 não foi descrita anteriormente em nenhum outro doente português com SG. Que tenhamos conhecimento, na literatura existe apenas duas descrições de doentes caucasianos com SG, que apresentam este alelo. O primeiro caso22 é o de uma rapariga de Taranto (Itália), heterozigota composta para uma e duas inserções ([TA]7/[TA]8), tal como o nosso caso. Este caso tem um grau elevado de semelhança com o nosso, já que são ambos do sexo feminino, apresentam a mesma idade e níveis de bilirrubina total muito semelhantes (60 e 57.9 μmol/L). Estes dois casos, analisados em conjunto dão alguma evidência da existência de algum tipo de correlação genótipo/fenótipo. O segundo caso,23 refere-se a um rapaz de 3 anos de idade de origem grega, heterozigoto para o alelo (TA)8 [(TA)6/(TA)8].
A presença do alelo (TA)8 é muito raro em indivíduos caucasianos, no entanto, a sua frequência na população africana é de 6.9 %. Iolascon e Tsezou22,23 sugerem que o aparecimento deste alelo na população caucasiana é um evento genético recente, não sendo portanto o resultado de uma mutação ancestral comum. Este raciocínio é reforçado pelo facto de sequências repetitivas serem instáveis e poderem aumentar ou diminuir por diferentes mecanismos, nomeadamente por crossing over desigual durante a meiose.
Icterícia neonatal
A sugestão de que o SG poderia influenciar o aparecimento e a gravidade da IN foi referida pela primeira vez em 1980. No entanto só quando foram identificadas alterações no gene UGT1A1 associadas ao SG, foi possível efectuar estudos que procuraram relacionar a IN e o SG. Relativamente a este assunto, nós estudamos24,25 100 recém-nascidos (RN) com IN (23 dos quais com IN prolongada), 100 RN com ausência de IN documentada e 18 doentes com diagnóstico de défice de glicose-6-fosfato desidrogenase (G6PD) com referência a IN, cinco dos quais com variantes associadas a anemia hemolítica não esferócitica crónica (AHNEC).
De entre os RN com IN, apenas o subgrupo (Quadro I) que desenvolveu IN prolongada apresentou uma proporção superior, estatisticamente significativa (χ2, p = 0.002), de homozigotia para o alelo (TA)7, quando comparado com o grupo controlo (39.1% versus 10%). Nos restantes RN, os nossos resultados sugerem que a presença deste polimorfismo não é um factor de risco major para o aparecimento da IN. No entanto, encontramos alguma evidência (apesar de não ser estatisticamente significativa dado o pequeno tamanho da amostra) de que a presença de homozigotia para a inserção TA em RN com outros factores de risco para desenvolvimento de IN condiciona um pico de bilirrubina total mais elevado (Figura 3).

Quadro I. Distribuição do genótipo do gene da UGT1A1 pelos RN estudados com e sem IN.

Figura 3. Comparação do pico de bilirrubina nos diferentes genótipos num grupo de RN ictéricos com outros factores de risco conhecidos.
Relativamente aos doentes com défice de G6PD que desenvolveram IN, encontramos uma incidência de homozigotia para variantes anormais do gene UGT1A1 apenas em 2 casos (11.11%). Esta proporção não é estatisticamente diferente da encontrada no grupo de RN sem IN documentada. Embora o número de doentes seja reduzido os nossos resultados sugerem que o aparecimento da IN em doentes com défice de G6PD é independente da associação com variantes anormais do gene do UGT1A1. Relativamente à gravidade da IN em doentes com variantes de G6PD relacionadas com AHNEC é de salientar que os 5 doentes incluídos (G6PDNara, G6PDDurham, G6PDTomah, G6PDAveiro e G6PDNashville) desenvolveram IN necessitando de transfusão permuta sem que, no entanto, nenhum deles apresentasse homo- ou heterozigotia para variantes do promotor do gene da UGT1A1. Concluímos que nesta situação a gravidade da icterícia neonatal resultará do aporte exagerado de bilirrubina resultante do processo hemolítico, mais do que da presença de variantes anormais do gene da UGT1A1.
Doença hemolítica crónica
A decisão de esplenectomizar um doente com DHC, nomeadamente com esferocitose hereditária, é controversa. Ao longo do tempo, a referência ao risco acrescido de sépsis grave em doentes esplenectomizados vem assumindo uma relevância progressiva, que se acentuou face à emergência de estirpes de pneumococo resistentes à penicilina. Como tal, e particularmente em crianças, tem vindo a ser feito um esforço no sentido de, ponderando a relação risco/benefício, objectivar o mais possível as suas indicações.4,5,17,26
Um dos eventuais benefícios da esplenectomia em doentes com DHC é o da prevenção da litíase vesicular.4,5 Como tal, tem vindo a ser feito um esforço no sentido de identificar doentes em risco acrescido de desenvolvimento desta complicação. Literatura recente inclui,5 por exemplo, a história familiar de litíase vesicular nos critérios de esplenectomia. A identificação a nível molecular de SG, em crianças com DHC, pode dar maior consistência à decisão delicada de proceder a uma esplenectomia em nome da prevenção da litíase vesicular. Isto mesmo em crianças cujo grau de hemólise não aparenta ser significativo.
Estudamos 50 crianças com DHC (esferocitose hereditária - 38, défice de glicose-6-fosfato desidrogenase com AHNEC - 5, défice de piruvato kinase – 6 e xerocitose hereditária - 1). Em todas elas, foram registados os valores médios de hemoglobina, reticulócitos e bilirrubina total. Foram excluídos os valores encontrados em fase de agudização. Nas crianças entretanto esplenectomizadas foram considerados apenas os valores referentes ao período pré esplenectomia. O diagnóstico de litíase vesicular foi efectuado por ultra-sonografia realizada anualmente de forma protocolada ou em contexto de sintomatologia dolorosa abdominal.27
Não encontramos diferença estatisticamente significativa no valor de hemoglobina e reticulócitos entre os três grupos de doentes (homozigóticos normais, heterozigóticos e hemozigóticos mutados), o que sugere um grau de hemólise similar entre os grupos. No entanto, relativamente aos valores de bilirrubina total (Figura 4) encontramos um aumento estatisticamente significativo nos doentes homozigóticos para a inserção dinucleotídica localizada na região TATA box do gene da UGT1A1 (teste de Mann-Whitney, p < 0.05). Neste último grupo encontramos um aumento também estatisticamente significativo, na proporção (Figura 5) de doentes que desenvolveram litíase vesicular (8% dos doentes com o genótipo [TA]6/[TA]6, 13.5 % dos doentes com o genótipo [TA]6/[TA]7 e 60% dos doentes com o genótipo [TA]7/[TA]7).
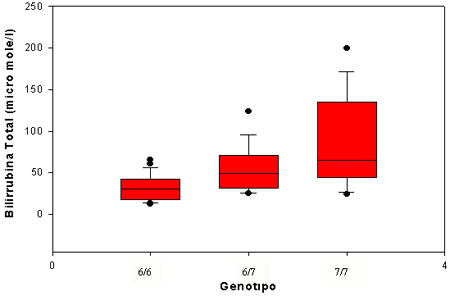
Figura 4. Distribuição dos valores de bilirrubina total nos 3 grupos de doentes.

Figura 5. Proporção de doentes que desenvolveram litíase vesicular e pancreatite aguda.
Quatro dos seis doentes com SG e que desenvolveram litíase vesicular, apresentaram pancreatite aguda como complicação da litíase vesicular, levantando a questão de se a associação de DHC e SG poderá aumentar não só o risco de litíase biliar, como também a gravidade das suas manifestações. Consequentemente, levantam ainda a questão da realização de esplenectomia como forma de prevenir não só a litíase vesicular como as suas complicações, mesmo nas situações em que a anemia é mínima ou ausente.
Conclusão
A pesquisa da inserção TA, efectuada por técnicas de biologia molecular simples, permite-nos estabelecer o diagnóstico de SG assim como fazer o estudo da sua influência na gravidade e morbilidade da hiperbilirrubinémia de outras situações clínicas com ela relacionadas. A análise dos nossos resultados sugere-nos que a presença do alelo [TA]7 é a principal responsável por SG na nossa população, está associada ao desenvolvimento de IN prolongada e, nos doentes com DHC, aumenta os níveis de bilirrubina e consequentemente o risco de desenvolvimento de litíase vesicular.
Los autores no manifiestan conflictos
Bibliografía del artículo
- Schmid R. Some aspects of the bile pigment metabolism. Clin Chem 1957;3:394-400.
- Schmid R. Direct-reacting bilirubin bilirubin glucuronide, in serum, bile, and urine. Science 1956;124:76-77.
- Biling BH, Cole PG, Lathe GH. The excretion of bilirubin as diglucuronide giving the direct van den Bergh reaction. Biochem J 1957;65:774-784.
- Lee GR, Bithell TC, Foerster J, Athens JW, Lukens JN. Wintrobe´s clinical haematology 9th ed. Lea and Febiger. Philadelphia 1993.
- Nathan DG, Orking SH. Nathan and Oski´s haematology of infancy and childhood 5th ed. WB Sanders Company. Philadelphia 1998.
- Bosma PJ, Chowdhury NR, Goldhoorn BG et al. Sequence of exons and the flanking regions of human bilirubin-UDP-glucuronosyltransferase gene complex and identification of a genetic mutation in a patient with Crigler-Najjar syndrome, type I. Hepatology 1992;15:941-7.
- Bosma PJ, Chowdhury JR, Bakker C, Gantla S, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert´s syndrome. N Engl J Med 1995;333:1171-5.
- Burchell B, Hume R. Molecular genetic basis of Gilbert´s syndrome. J Gastroenterol Hepatol 1999; 14:960-966.
- Beutler E, Gelbart T, Demina A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism Proc Natl Acad Sci USA 1998; 95:8170-4.
- Sampietro M, Iolascon A. Molecular pathology of Crigler-Najjar type I and II and Gilbert´s syndrome. Haematologica 1999;84:150-157.
- Biondi ML, Turri O, Dilillo D, Stival G, Guagnellini E. Contribution of the TATA-box genotype (Gilbert syndrome) to serum bilirubin concentrations in Italian population. Clinical Chemistry 1999;45:897-8.
- Pirulli D, Giordano M, Puzzer D, Crovella S, Rigato I, Tiribelli C, et al. Rapid method for detection of extra (TA) in the promoter of the bilirubin-UDP-glucuronosyl transferase 1 gene associated with Gilbert syndrome. Clinical Chemistry 2000;46:129-31.
- Kaplan M, Hammarman C, Renbaum P, Klein G, Levy-Lahad E. Gilbert´s syndrome and hyperbilirubinaemia in ABO-incompatible. Lancet 2000; 356:652-3.
- Kaplan M, Beutler E, Vreman HJ, Harmmerman C, Levy-Lahad E, Renbaum P, Stevenson DK. Neonatal hyperbilirubinemia in glucose-6-phosphate dehydrogenase deficient heterozygotes. Pediatrics 1999; 104:68-74.
- Iolascon A, Faienza MF, Giordani L, Perrotta S, Ruggiu G, Meloni GF, del Giudice EM. Bilirrubin levels in the acute hemolytic crisis of G6PD deficiency are related to Gilbert´s syndrome. Eur J Haematol 1999; 62:307-10.
- Galanello R, Perseu L, Melis MA, Cipollina L, Barella S, Giagu N, Turco MP, Maccioni O, Cao A. Hyperbilirubinaemia in heterozygous beta-talassaemia is related to co-inherited Gilbert´s syndrome. Br J Haematol 1997;99:433-436.
- Giudice EM, Perrota S, Nobili B, Specchia C, d´Urzo G, Iolascon A. Coinheritance of Gilbert syndrome increases the risk for developing gallstones in patients with hereditary spherocytosis. Blood 1999;94:2259-62.
- Bancroft JD, Kreamer B, Gourley GR. Gilbert syndrome accelerates development of neonatal jaundice. J Pediatr 1998; 132:656-660.
- Passon RG, Howard TA, Zimmerman SA, Schultz WH, Ware RE. Influence of bilirubin uridine diphosphate glucuronosyltransferase 1A promoter polymorphisms on serum bilirubin levels and colelithiasis in children with sickle cell anemia. J Pediatr Hematol Oncol 2001;23:448-451.
- Borgna-Pignatti C, Rigon F, Merlo L, et al. Thalassemia minor, the Gilbert mutation, and risk of gallstones. Haematologica 2003;88:1106-1109.
- Costa E, Vieira E, Santos-Siva E, et al. TATA-box polymorphism in the uridine diphosphate glucuronosyl transferase gene in Portuguese patients with a clinical diagnosis of Gilbert's syndrome. Haematologica 2002; 87:(04)ELT21.
- Iolascon A, Faienza MF, Centra M, Storelli S, et al. (TA)8 allele in the UGT1A1 gene promoter of a caucasian with Gilbert´s syndrome. Haematologica 1999;84:106-109.
- Tsezou A, Tzetis M, Kitsiou S, et al. A Caucasian boy with Gilbet´s syndrome heterozygous for the (TA)8 allele. Haematologica 2000;85:319.
- Alexandrino AM, Carvalho C, Costa E, Vieira E, Oliveira P, Duarte C, Barbot J, dos Santos R, Areias A. TATA box polymorphism in the UDP-glucoronosyltransferase 1 gene promoter and neonatal hyperbilirubinemia. Prenatal and Neonatal Medicine 2001; 6:133-136.
- Costa E, Vieira E, Cleto E, et al. Glucose-6-phosphate dehydrogenase deficiency, neonatal hyperbilirubinemia and Gilbert syndrome. Acta Med Port 2002;15:409-12.
- Schilling RF. Sherocytosis, splenectomy, stroke, and heart attacks. Lancet 1997;350:1677-78.
- Costa E, Pinto R, Vieira E, et al. Influence of Gilbert's syndrome on serum bilirubin levels and gallstone formation in children with chronic hemolytic disease An Esp Pediatr 2002;57:529-33.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC



