EXPOSIÇÃO AOS POLUENTES DA QUEIMA DE CANAVIAIS E O RISCO PARA SAÚDE
(especial para SIIC © Derechos reservados)
Coautor
Nívea Conforti-Froes.*
Doutor em Genética, Pós-Doutor em Medicina Preventiva. Universidade Estadual Paulista (UNESP). Instituto de Biociencias, Letras e Ciencias Exatas, campus de Sao José do Rio Preto.*
Recepción del artículo: 21 de abril, 2004
Aprobación: 3 de agosto, 2004
 Conclusión breve
Esta revisão aborda a interação entre o ambiente e a herança genética no aparecimento do câncer, assim como apresenta as ferramentas disponíveis para a detecção do risco de exposição a esses compostos, facilitando a aplicação de estratégias de prevenção da doença.
Resumen
No Brasil, na época da colheita da cana-de-açúcar, costuma-se queimar previamente a plantação para facilitar o corte. No período da safra, os cortadores de cana estão expostos a vários produtos originados da combustão de matéria orgânica, incluindo os hidrocarbonetos aromáticos policíclicos (polycyclic aromatic hydrocarbons [PAHs]), compostos já conhecidos por sua ação mutagênica e/ou carcinogênica. O cortadores de cana também apresentam um aumento de problemas respiratórios, possivelmente resultante da exposição aos poluentes originados da queima de canaviais. Muitas das enzimas envolvidas no processos de ativação e destoxificação dos PAHs são polimórficas. Os polimorfismos genéticos podem desempenhar um papel importante no aumento da suscetibilidade a tumores induzidos por xenobióticos. Esta revisão aborda a interação entre o ambiente e a herança genética no aparecimento do câncer, assim como apresenta as ferramentas disponíveis para a detecção do risco de exposição a esses compostos, facilitando a aplicação de estratégias de prevenção da doença.
Palabras clave
Hidrocarbonetos aromáticos policíclicos, cana-de-açúcar queimada, polimorfismos genéticos, poluentes ambientais, estratégias de prevenção do câncer
Clasificación en siicsalud
Conclusión breve
Esta revisão aborda a interação entre o ambiente e a herança genética no aparecimento do câncer, assim como apresenta as ferramentas disponíveis para a detecção do risco de exposição a esses compostos, facilitando a aplicação de estratégias de prevenção da doença.
Resumen
No Brasil, na época da colheita da cana-de-açúcar, costuma-se queimar previamente a plantação para facilitar o corte. No período da safra, os cortadores de cana estão expostos a vários produtos originados da combustão de matéria orgânica, incluindo os hidrocarbonetos aromáticos policíclicos (polycyclic aromatic hydrocarbons [PAHs]), compostos já conhecidos por sua ação mutagênica e/ou carcinogênica. O cortadores de cana também apresentam um aumento de problemas respiratórios, possivelmente resultante da exposição aos poluentes originados da queima de canaviais. Muitas das enzimas envolvidas no processos de ativação e destoxificação dos PAHs são polimórficas. Os polimorfismos genéticos podem desempenhar um papel importante no aumento da suscetibilidade a tumores induzidos por xenobióticos. Esta revisão aborda a interação entre o ambiente e a herança genética no aparecimento do câncer, assim como apresenta as ferramentas disponíveis para a detecção do risco de exposição a esses compostos, facilitando a aplicação de estratégias de prevenção da doença.
Palabras clave
Hidrocarbonetos aromáticos policíclicos, cana-de-açúcar queimada, polimorfismos genéticos, poluentes ambientais, estratégias de prevenção do câncer
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/68038
Especialidades
 Principal: Medicina del Trabajo, Oncología,
Principal: Medicina del Trabajo, Oncología,
Relacionadas: Bioquímica, Epidemiología, Genética Humana, Salud Pública,
Enviar correspondencia a:
Rosa Maria do Vale Bosso. Cristóvao Colombo, n 2265-Bairro, Jd. Nazareth, Sao José do Rio Preto, Sao Paulo- Brasil. CEP: 15054-000 Bosso, Rosa Maria
EXPOSURE TO POLLUTANTS OF SUGAR CANE PLANTATION BURNT AND HEALTH RISK
Abstract
In Brazil, the leaves of the sugar cane are burnt before harvesting in order to make the process of cutting easier. During the harvest time, the sugar cane workers are exposed to various combustion products of organic materials including polycyclic aromatic hydrocarbons (PAHs) which are known to be mutagenic and/or carcinogenic. The workers also have an increased risk of lung respiratory disorders, possibly due to pollutants exposure derived from the practice of burning foliage during the cane-cutting time. Many of the enzymes involved in the activation and detoxification processes of PAHs are genetically polymorphic. Genetic polymorphims can play an important role in increasing susceptibility to xenobiotic-induced tumors. This review approaches the interaction between environmental pollutants and genetic inheritance in the carcinogenic process, as well as the current available tools for risk detection of environmental exposure, thus enabling the application of cancer-prevention public strategies.
Key words
Polycyclic aromatic hydrocarbons (PAHs), sugar cane burnt, genetic polymorphism, environmental pollutants, cancer prevention strategies
EXPOSIÇÃO AOS POLUENTES DA QUEIMA DE CANAVIAIS E O RISCO PARA SAÚDE
(especial para SIIC © Derechos reservados)
Artículo completo
Introdução
O Brasil é o maior produtor de cana-de-açúcar do mundo, seguido pela Índia e Austrália. Aproximadamente, 55% da cana brasileira transformam-se em álcool e 45%, em açúcar. A cana é plantada no Centro-Sul e no Norte-Nordeste, permitindo dois períodos de safra, portanto, sua produção é observada o ano todo. Em vista disso, na década de 70, foi implantado o PROALCOOL, um programa desenvolvido pelo governo brasileiro, com a finalidade de incentivar a utilização do álcool como combustível automobilístico. A partir de então, o Brasil, sustentado pela agroindústria canavieira, tem sido o pioneiro na utilização desse combustível limpo e renovável. Dentre os estados brasileiros, o de São Paulo gera 61% da produção brasileira de álcool, permitindo ao país continuada economia de divisas, em um ritmo médio de 1.8 bilhões de dólares anuais. De modo similar, esse estado produz 60% de todo o açúcar nacional, que por ser produto competitivo no mercado internacional, torna o estado responsável por 70% das exportações. Além disso, da cana-de-açúcar origina-se a energia elétrica, co-gerada no processo de queima do bagaço.1
Para produzir açúcar e álcool, a área de cana colhida no Estado de São Paulo, na safra 2003/2004, foi de aproximadamente 2.45 milhões de hectares, o que proporcionou o emprego de cerca de 400 mil pessoas no campo e nas usinas. Isso significa 40% do trabalho rural do Estado.2
No Brasil, para facilitar o corte da cana-de-açúcar, costuma-se queimar previamente os canaviais. Desse processo, origina-se uma fuligem que permanece por um tempo no ar e tem sido apontada, pelos próprios cortadores de cana, como responsável pelo aumento de problemas respiratórios. Além desse fato, os poluentes originados dessa operação, são levados de acordo com a direção do vento, contaminando não apenas os trabalhadores rurais, mas o ambiente como um todo, que inclui os moradores de municípios vizinhos.
Do processo de queima de canaviais são formados, dentre outros poluentes, os hidrocarbonetos aromáticos policíclicos (polycyclic aromatic hydrocarbons [PAHs]), compostos conhecidos por sua ação mutagênica e/ou carcinogênica, ocorrendo primariamente como resultado da combustão e pirólise, a partir de matéria orgânica.3
Os PAHs são moléculas orgânicas formadas por átomos de hidrogênio e carbono. A maior fonte de tais compostos compreende a emissão de materiais particulados em conseqüência da queima de plantações agrícolas, da fumaça do cigarro, da combustão do carvão, dos motores de veículos e da produção de coque.4,5 A combustão é um processo químico de oxidação que ocorre em uma taxa suficientemente rápida para produzir elevação da temperatura originando calor ou chama.6 Desse modo, são compostos encontrados no ambiente ocupacional e residencial e, em conseqüência da dispersão das cinzas, atinge o homem via inalação, dérmica ou pela água de rios contaminados.7 Representam uma classe importante de poluentes ambientais, pois as lesões resultantes das ligações desses compostos com o DNA, quando não reparadas, poderão iniciar o processo carcinogênico.8
A Enviromental Protection Agency (EPA) e National Institute of Occupational Safety and Health (NIOSH),9,10 dos Estados Unidos, consideram os PAHs como poluentes prioritários para investigação ambiental, cujas estruturas estão representadas na figura 1. Essas duas agências do governo norte-americano estão sendo citadas, uma vez que no Brasil não há legislação ambiental e/ou ocupacional, em relação aos PAHs.
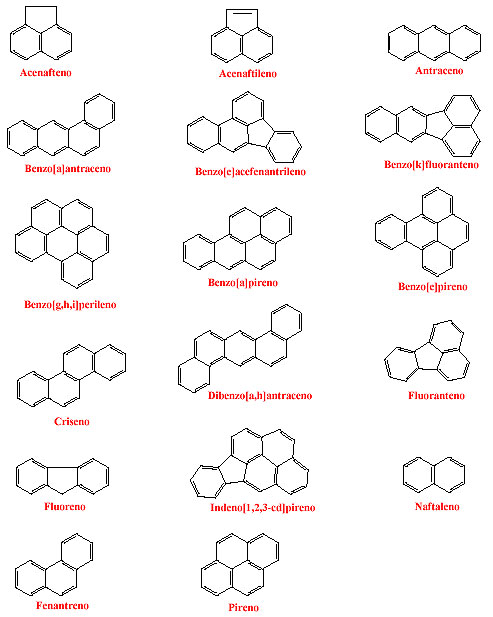
Desde 1920, uma intensa série de investigações tem sido empreendida visando identificar e isolar os PAHs com atividade carcinogênica. Um dos mais conhecidos, o benzo(a)pireno [B(a)P], está presente em quantidade considerável na fumaça do cigarro e foi reconhecido como seu principal e mais ativo componente.11
A partir dos estudos de Miller & Miller,12 houve a expansão do conhecimento sobre o metabolismo dos compostos químicos, estabelecendo-se que esses compostos não são ativos na forma em que são ingeridos ou inalados, tornando-se reativos apenas após serem convertidos enzimaticamente, por meio de reações específicas do metabolismo. Em vista disso, pode-se esclarecer os passos envolvidos no metabolismo dos PAHs.
Resumidamente, seu metabolismo é mediado pela aril–hidrocarboneto-hidroxilase (AHH), uma das muitas monoxigenases de função mista que participam das reações de ativação metabólica. Uma vez em contato com o organismo, esses compostos poderão seguir duas vias que incluem a da bioativação ou da bioinativação. Nessa última, serão transformados, pela ação da glutatião e glicuronil transferases, em produtos conjugados, com pouca ou nenhuma oportunidade de reagirem com o DNA, sendo facilmente excretados. Entretanto, se os mesmos forem hidratados a dihidrodiol, pela ação da epóxido hidratase, e sofrerem uma nova oxidação, pela ação da citocromo P-450 monoxigenase, darão origem ao diidrodiol epóxido, forma genotóxica final do composto e altamente reativa ao DNA.13-15 A figura 2 ilustra essas vias alternativas, tomando-se como exemplo o B(a)P.
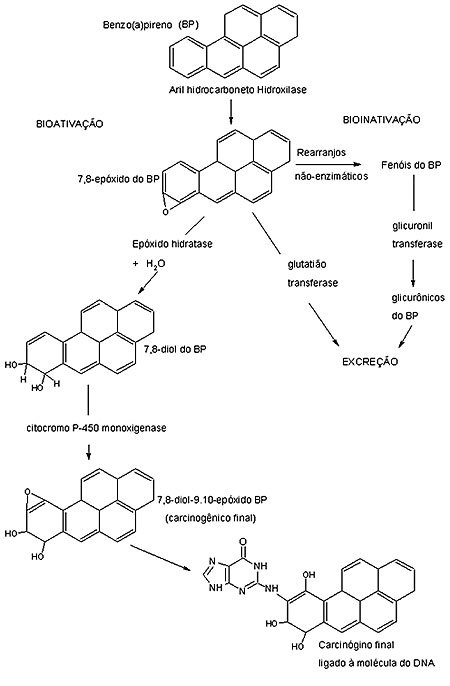
No que se refere às ligações dos metabólitos PAHs ao DNA, as reações ocorrem, predominantemente, no grupo amino exocíclico das bases nitrogenadas.16 Há muitos anos, sabe-se que mais de 90% da ligação de um radical alquil ao DNA, causadas pela forma genotóxica final originada do metabolismo do B(a)P, ou seja, o 7,8-dihidrodiol 9,10-epóxido (BPDE), ocorrem no grupo amino exocíclico da guanina. Outros sítios de alquilação incluem o oxigênio na posição 6, o nitrogênio na posição 7 da guanina, além do grupo amino exocíclico da adenina e citosina.17 Canella et al.18 demonstraram que a ligação do BPDE com o DNA resulta de uma abertura trans do anel de epóxido pelo grupo amino exocíclico dos resíduos da desoxiguanosina. Existe uma significativa diferença na ligação de determinados resíduos de PAHs com o DNA. Essa diferença na reatividade deve-se à estrutura química desses compostos. Por exemplo, o dimetil-benzo(a)antraceno (DMBA) possui um grupo metil que permite sua ligação tanto com a desoxiadenosina, bem como a desoxiguanosina.15 Essas diferenças de conformação podem, presumivelmente, levar a diferenças nas vias em que os adutos de DNA são reparados ou não durante o processo da duplicação celular, conferindo capacidade diferencial na indução de mutações gênicas e no aparecimento de uma lesão pré-neoplásica irreversível.15
Assim, pode-se depreender que, a extensão dos danos ao DNA, causados pelos PAHs, parece estar intimamente relacionada com a conformação estrutural da molécula, às reações metabólicas a que são submetidos esses compostos, bem como ao tipo de metabólitos formados que, quando em suas formas finais, atingem os órgãos-alvo, levando a formação de lesões no material genético, que são, reconhecidamente, fatores de risco para o desenvolvimento do câncer humano.19
Classificação dos agentes químicos quanto ao potencial tóxico
Os agentes químicos são classificados em grupo, segundo seu potencial tóxico, de acordo com o International Agency for Research on Cancer.20,21 No grupo 1, o agente tóxico é provavelmente carcinogênico, existindo evidências suficientes de carcinogenicidade em estudos que incluem o homem (estudos epidemiológicos). No grupo 2, o agente tóxico é provavelmente carcinogênico. Esse grupo compreende dois subgrupos, sendo o 2A, aquele em que existem certificações limitadas de carcinogenicidade em seres humanos e suficientes em animais de laboratório. O 2B é usado quando existem evidências inadequadas ou inexistência de carcinogenicidade em relação ao homem e constatações suficientes em animais de laboratório. No grupo 3, o agente tóxico é possivelmente carcinogênico para o homem. Nesse grupo, estão incluídos os compostos com evidências limitadas em estudos com animais de laboratório e ausência de evidências no homem. Em relação ao grupo 4, o agente tóxico não pode ser classificado em função da ausência de informações ou pela existência de estudos, no homem e em animais, com evidências inadequadas. No grupo 5, o composto tóxico não é carcinogênico, existindo estudos que comprovam a ausência de efeitos ao homem e animais de laboratório.
Na tabela I estão as características mutagênicas e carcinogênicas dos 17 PAHs prioritários pela EPA e NIOSH e sua classificação de acordo com a IARC.22,20
Tabela 1Os PAHs e o câncer
Durante séculos, houve o predomínio da idéia de que a natureza existia somente para satisfazer as vontades humanas, sem se questionar o limite desse usufruto. Embora muitos ainda pensem dessa maneira, a cada dia vêem-se sinais de que a sociedade está se conscientizando sobre os danos irreversíveis que a poluição, provocada pelo homem, pode causar ao ambiente e ao próprio indivíduo.
O reconhecimento das propriedades carcinogênicas dos compostos originados do processo de combustão é muito antigo, pois em 1775, um cirurgião inglês, Sir Percival Pott, atribuiu a alta incidência de câncer de testículo e da cavidade nasal observada em limpadores de chaminés à prolongada exposição desses indivíduos à fuligem. A observação de Pott foi a base para a primeira medida preventiva contra o câncer ambiental, já que pouco depois de sua publicação, a associação dinamarquesa dos limpadores de chaminés passou a estimular seus membros a tomarem banhos diários e, em 1892, notou-se uma queda na incidência desse tipo de câncer nesses trabalhadores.23
A existência de uma íntima relação entre os processos mutagênico e carcinogênico foi primeiramente sugerida por Theodor Boveri, em 1914, que propôs ser o câncer dependente da ocorrência de um evento mutacional. Na verdade, sabe-se hoje que a progressão neoplásica da maioria dos cânceres humanos comuns ocorre como resultado de eventos mutacionais múltiplos, sucedidos por alterações bioquímicas e fenotípicas das células.24 Esse processo afeta, de forma não controlada, a diferenciação e a proliferação celular. Embora na sua fase inicial a neoplasia seja localizada, pode disseminar-se pelo organismo em conseqüência da capacidade das células cancerosas crescerem em outros tecidos, o que pode ser letal para o indivíduo.25
Consideram-se, pelo menos, três etapas nesse processo, desde o momento da agressão química até o aparecimento da neoplasia. Os primeiros eventos representam o passo em que o agente externo é processado metabolicamente pelo organismo e liga-se a macromoléculas como o DNA. É seguida por um ciclo de divisão celular, em que a lesão bioquímica é fixada e transforma as células-alvo em células iniciadas ou mutadas. A característica da "iniciação" é que as alterações ainda podem ser reparadas. Na segunda etapa, designada "promoção", existe uma lesão pré-neoplásica. Ocorre a inativação de genes supressores de tumor e ativação de proto-oncogenes. A "progressão", terceira etapa da carcinogênese, é entendida como o passo em que podem ocorrer alterações qualitativas no fenótipo celular, levando à transformação de uma lesão benigna em maligna, possivelmente por seleção clonal.26
O tempo entre o efeito iniciador e o aparecimento de um tumor é denominado período de latência e, no homem, parece variar de 10 a 20 anos. Enquanto o acúmulo de mudanças genéticas em células somáticas é considerado essencial para a gênese do câncer, sabe-se que nem todo carcinógeno é genotóxico, podendo participar de modo indireto como promotor durante a carcinogênese.27
O câncer é um sério problema de saúde pública. Atualmente, é responsável por cerca de dez milhões de novos casos anuais, com seis milhões de mortes a cada ano no mundo. Desse total, 50% dos casos concentram-se em países em desenvolvimento, que disponibilizam apenas 5% de seus recursos para o controle da doença. Caso não haja uma estratégia efetiva de prevenção, detecção e cuidados, no ano de 2020 serão vinte milhões de novos pacientes/ano com a doença, estimando-se uma mortalidade anual de doze milhões.28
Dados epidemiológicos indicam que a maioria dos cânceres ocorre devido a fatores ambientais. Conseqüentemente, a identificação de carcinógenos é de fundamental importância para a prevenção da doença, sendo os poluentes, como os pesticidas sintéticos e outros compostos químicos produzidos pelo homem, um risco importante de câncer para a população geral.29 Realmente, existe forte correlação entre exposição a altas concentrações de PAHs e incidência de câncer de pulmão,30 esôfago, cabeça e pescoço, cólon e bexiga, que estão ligados, por sua vez, também ao uso do tabaco.31-34
Vários estudos têm mostrado que muitos PAHs são mutagênicos em células bacterianas e em células de cultura de mamíferos, além de induzirem aberrações cromossômicas (AC) e elevarem a freqüência de micronúcleos (MN) e de trocas de segmentos entre cromátide irmãs (TCI).35,36 Esses parâmetros são importantes sinais pré-clínicos de uma doença e suas freqüências aumentadas estão diretamente relacionados com a incidência aumentada de doenças, como o câncer.A avaliação de amostras de ar das cidades de São Paulo, Cubatão37 e Porto Alegre,38 utilizando o teste para avaliação de mutagenicidade com Salmonella typhimurium, sugeriu a presença de nitro-PAHs nesses locais. Além da atmosfera nas grandes cidades e nas indústrias, que contêm altos níveis de poluentes, outros locais têm revelado a presença desses compostos. Zamperlini et al.,39 por exemplo, utilizando técnicas cromatográficas e espectrofotométricas, detectaram PAHs (do tipo considerado poluente prioritário) na fuligem originada da queima de canaviais. Por meio do teste de Ames, foi demonstrada atividade mutagênica na fuligem sedimentada da cana-de-açúcar, além de índices mais elevados de mutagenicidade em amostras de urina de cortadores de cana, quando comparadas às de controles.40 Amre et al.41 observaram um risco elevado de câncer de pulmão em trabalhadores rurais de plantações de cana-de-açúcar. Os dados apresentados mostram que a população está exposta a complexas misturas de compostos químicos, principalmente aquelas ricas em PAHs.
Polimorfismos e correlação entre marcadores de exposição e suscetibilidade
Os marcadores biológicos são indicadores do risco genotóxico e podem ser usados para a detecção precoce de populações de risco, bem como agentes danosos presentes no ambiente. São subdivididos em indicadores de exposição, efeito e de suscetibilidade. De acordo com essa classificação, é possível identificar um indicador biológico em cada passo da substância genotóxica dentro do organismo, desde sua absorção, ataque nucleofílico e conseqüente lesão às moléculas informacionais, fixação do dano e excreção.
No monitoramento biológico deve ser feita a distinção entre indicadores de exposição e os de efeito. O primeiro inclui a concentração da substância ou seus metabólitos em fluidos biológicos, a mutagenicidade da substância excretada e o ataque metabólico ao DNA. Tais indicadores têm o propósito de avaliar a absorção do composto em questão e se possível, sua dose interna biologicamente efetiva, que refere-se à sua concentração no organismo. Os indicadores de efeito incluem a detecção de sinais pré- clínicos ou clínicos da doença, como o aumento na freqüência de aberrações cromossômicas, a mutação em oncogenes e genes supressores de tumor, que fornecem o grau das alterações de alvos genéticos importantes, como em cromossomos e seqüência gênica. Por outro lado, os marcadores de suscetibilidade incluem as diferenças individuais no metabolismo de xenobióticos, no reparo de DNA e a presença de polimorfismos em oncogenes e genes supressores de tumor. Esses são, atualmente, de grande interesse no campo da medicina ambiental, principalmente devido ao seu papel na modulação da resposta individual à exposição a carcinógenos.42 Para efeito ilustrativo, abordaremos um exemplo em que biomarcadores de exposição e suscetibilidade são combinados, para melhor esclarecer o intricado processo do metabolismo de xenobióticos e risco à populações expostas. A quantificação do 1-hidroxipireno (1-OHPyr) que é um metabólito do pireno será o exemplo utilizado como indicador biológico da dose interna (biomarcador de exposição) na avaliação da exposição aos PAHs. Sua excreção representa 90% da metabolização do pireno, que é encontrado em 1% a 10% nas misturas de PAHs.43 O 1-OHPyr é excretado na urina ligado ao ácido glicurônico, ao contrário dos outros metabólitos dos PAHs, que são excretados principalmente nas fezes.44 Entretanto, cerca de 10% a 25% foram encontrados na urina em forma livre.45 A utilização do 1-OHPyr no monitoramento biológico da exposição aos PAHs é vantajosa, devido ao fato do pireno estar presente nas misturas de PAHs em concentrações relativamente altas, ser estável, possuir apenas o pireno como precursor, passível de determinação em urina humana, incluindo populações controles.46 Além disso, há correlação entre as concentrações do pireno com o benzo(a)pireno e outros PAHs, sendo assim considerado um bom indicador da exposição aos PAHs.22
Ao entrar em contato com os PAHs, os indivíduos respondem diferentemente. Isso se deve ao fato de que a maioria dos compostos químicos não é ativa por si só, necessitando de ativação metabólica para tornar-se eletrofílica e interagir-se com as macromoléculas celulares.47 Desse processo, decorre a necrose tecidual e a carcinogenicidade. Assim sendo, a variação no fenótipo metabólico, que envolve as enzimas de ativação e destoxificação, desempenha um papel importante no desenvolvimento do câncer ambiental. Essa variação está relacionada ao polimorfismo genético, sendo que grande parte desse polimorfismo apresenta relevância clínica e envolve as enzimas metabolizadoras de carcinógenos.48 Tais enzimas são didaticamente classificadas em duas amplas categorias, as de fase I e as de fase II. As enzimas da fase I envolvem quase que exclusivamente os citocromos P-450 (CYP-450), que catalisam a inserção de um átomo de oxigênio em substratos relativamente inertes, tornando-os altamente reativos. Por outro lado, as enzimas de fase II, que incluem principalmente as glutationas S-transferases (GSTs), reagem com os eletrofílicos finais, que se tornaram reativos na fase I, catalisando a conjugação desses com substratos endógenos, como glicuronídeos, glutationas e sulfatos. Consequentemente, são produzidos intermediários hidrofílicos, facilmente excretáveis da célula, embora tais intermediários também possam ser originados por um processo não enzimático.49
O gene que codifica a isoenzima humana CYP1A1 localiza-se no braço longo do cromossomo 15, possuindo sete éxons e seis íntrons.50 A enzima CYP1A1 é induzida principalmente em tecidos extra-hepáticos, pelos vários compostos que possuem capacidade de se ligar ao receptor aril hidroxilase (Ah), responsável pela regulação do gene CYP1A1. Entre esses compostos, estão incluídas substâncias como dioxinas, benzoapireno (BaP) e outros PAHs. Ao se ligarem ao receptor Ah no citosol, o complexo é conduzido ao núcleo e a transcrição do gene CYP1A1 é induzida. O produto gênico do CYP1A1 possui atividade aril hidrocarboneto hidroxilase (AHH) que catalisa o primeiro passo do metabolismo dos PAHs a compostos eletrofílicos, podendo culminar em metabólitos carcinogênicos.51
Até o momento onze polimorfismos do CYP1A1 foram descritos,52 no entanto, dois deles vêm sendo mais estudados. A freqüência de cada um depende do grupo étnico.53 O polimorfismo, denominado atualmente por CYP1A1*2A, foi localizado na posição 3801, 264 bases após o sinal de poliadenilação, na região 3' que flanqueia o gene e 1194 pares de bases após o término do éxon 7. A transição de timina para citosina dá origem a um sítio de restrição para a enzima MspI.54 O segundo, foi localizado na região codificadora do éxon 7 e é representado pela transição da adenina para guanina na posição 2455.55 O polimorfismo no éxon 7, também denominado CYP1A1*2B, leva a uma mudança do aminoácido Isoleucina para Valina (CYP1A1 Ile/Val) no resíduo 462 na região de ligação ao grupamento heme (HR2). Uma cisteína localizada anteriormente na posição 457 faz parte da ligação com o grupamento heme. Desse modo, esse polimorfismo está localizado em uma região essencial para a atividade enzimática e a mudança de aminoácidos poderia resultar em alteração de atividade.55
Dentre esses dois polimorfismos, o CYP1A1*2A é o mais freqüente. Os povos de origem asiática apresentam maior freqüência alélica (0.37), seguida dos africanos (0.24), latinos (0.20) e caucasianos (0.12). Os asiáticos também apresentam maior freqüência em relação ao polimorfismo CYP1A1*2B (0.22), seguidos pelos latinos (0.16) e caucasianos (0.09) sendo que esse polimorfismo não está presente em africanos. Nos indivíduos de origem hispânica, ambos os polimorfismos apresentam freqüência intermediária, quando se compara caucasianos e asiáticos.53 Nessas populações existe uma ligação entre os polimorfismos CYP1A1*2B e o CYP1A1*2A, ou seja, os dois ocorrem simultaneamente, caracterizando-se um desequilíbrio de ligação. Tal ligação não foi observada na população afro-americanana, ao contrário dos achados de Hamada et al.,56 em população brasileira da cidade do Rio de Janeiro.
As glutatião S-transferases (GSTs) podem ser encontradas na maioria dos organismos vivos, incluindo as bactérias, fungos e plantas. Uma grande quantidade dessas enzimas se expressa nos organismos superiores, como em humanos.49 Dentre elas encontram-se, pelo menos, sete classes, das quais duas estão ligadas à membrana, cinco delas são citosólicas e incluem as alfa (α), mi (μ), pi (π), theta (θ) e kappa (κ). Apesar disso, as mais estudadas são as θ (GSTT1), π (GSTP1) e μ (GSTM1).57,51
A deleção homozigota do gene GSTT1, isto é, o genótipo nulo (GSTT1 0/0) apresenta uma freqüência que varia de 9.7% a 38% na população geral, dependendo da origem étnica. Foi observado que a ausência da atividade enzimática aumenta o risco para câncer de cabeça e pescoço, laringe, bexiga e pulmão.58,59
Os polimorfismos do gene GSTP1 constituem duas variantes alélicas, a GSTP1*B e GSTP1*C, sendo a GSTP1*A o tipo selvagem. A GSTP1*B difere da GSTP1*A pela transição A>G no nucleotídio 313, códon 104: Ile>Val e a GSTP1*C pela transição C>T no nucleotídeo 341, códon 113: Val > Ala. Alguns autores relataram uma freqüência aumentada de GSTP1*B em diferentes pacientes, homozigotos, com câncer de pulmão, oro-faringe, carcinoma de laringe e algumas outras doenças relacionadas à fumaça de cigarro.59
A classe μ inclui, pelo menos, 5 genes que codificam para um número igual de enzimas (GSTM1, M2, M3, M4, M5). O gene que codifica para a isoforma GSTM1 é polimórfico e tem 4 variantes alélicas incluindo, GSTM1*A, *B, *C, e *0. Os dois primeiros não apresentam diferenças com relação ao tipo de substrato, o alelo *C é extremamente raro e a variante *0, alelo nulo, acarreta a falta da atividade enzimática quando em homozigose.42 Dessa maneira, a presença de, pelo menos, um alelo funcional é suficiente para que a enzima atue desempenhando seu papel. A enzima GSTM1 se expressa em vários tecidos incluindo o fígado, rins, bexiga, estômago, leucócitos e em menor extensão nos pulmões, sendo responsável pela inativação de uma variedade de intermediários reativos, tais como os derivados dos PAHs.60
O genótipo nulo acomete cerca de 50% da população caucasóide51 e apresenta uma variação entre 30% a 70% para diferentes grupos étnicos.61,62 Os resultados obtidos em nosso laboratório mostraram freqüências similares entre populações das regiões Sudeste e Norte do Brasil para os genótipos homozigotos nulos dos genes GSTM1/T1. Tais freqüências são respectivamente de 47.3% e 18.5% em ambas as populações,63 não diferindo dos valores descritos para a população caucasóide de diferentes países, conforme citado anteriormente.
A correlação de biomarcadores de suscetibilidade e de exposição é bastante eficiente no estabelecimento de estratégias preventivas. Contudo, apenas alguns estudos avaliaram a influência dos polimorfismos metabólicos na atividade mutagênica da urina em indivíduos ocupacional ou ambientalmente expostos aos compostos genotóxicos. O trabalho de Hirvonen et al.64 mostrou que indivíduos fumantes que herdaram genótipo GSTM1 nulo apresentavam maior mutagenicidade na urina, comparados aos indivíduos fumantes com genótipo GSTM1 positivo. Binkovà et al.65 amostraram mulheres de uma área altamente poluída na Polônia, com a finalidade de verificar exposição ambiental a PAHs, encontrando atividade mutagênica urinária aumentada no grupo que apresentou GSTM1 nulo. Gabanni et al.66 mostraram que indivíduos fumantes, trabalhadores de fornos de coqueria, que possuíam fenótipo acetilador lento, ou seja que produzem proteínas que são instáveis, pobremente expressadas ou que têm atividade catalítica parcialmente reduzida, associado ao genótipo GSTM1 nulo apresentavam maior mutagenicidade da urina, comparados aos indivíduos que possuíam o genótipo normal.
Já em relação à exposição terapêutica, foi observado que a urina de pacientes não fumantes, com genótipo GSTM1 nulo e portadores de psoríase, tratados topicamente com ungüento contendo 2% de alcatrão de carvão, era duas vezes mais mutagênica que a urina de indivíduos portadores do genótipo normal.67
Vários estudos têm avaliado a influência dos polimorfismos genéticos nos níveis de 1-OHPyr urinário nas populações alvo. O trabalho de Binkovà et al.65 que avaliou a exposição ambiental em mulheres não fumantes também quantificou os metabólitos urinários de alguns PAHs, tais como pireno, benzoapireno, criseno e encontraram um valor médio duas vezes maior em indivíduos GSTM1 nulo, quando comparados com o grupo GSTM1 positivo. Entretanto, em outro trabalho na mesma população feminina, os níveis urinários dos metabólitos dos PAHs correlacionaram-se melhor com os níveis de exposição ambiental em indivíduos NAT2 lento e GSTM1 nulo.68 Em oposição às expectativas, Hemminki et al.69 não encontraram influência dos polimorfismos CYP1A1*2A, *2B e GSTM1 nos níveis de 1-OHPyr urinário em indivíduos que trabalham com fundição. Merlo et al.70 avaliando policiais do tráfico de cidade de grande porte, não detectaram qualquer influência do genótipo CYP1A1, GSTM1 e GSTT1 nos níveis de 1-OHPyr, embora a excreção tenha sido maior em alguns fumantes (≤ 15 cigarros/dia) com genótipo heterozigoto CYP1A1-MspI (CYP1A1*1/*2A), comparados aos homozigotos normais (CYP1A1*1/*1). Pavanello et al.71 avaliando indivíduos expostos a PAHs, como trabalhadores de fornos de coqueria, de indústrias de alumínio e limpadores de chaminé, demonstraram que, dentro de cada grupo, os valores médios de 1-OHPyr foram maiores em indivíduos GSTM1 nulo em relação ao grupo GSTM1 positivo, embora a diferença não tenha sido estatisticamente significante. Ao contrário dos estudos anteriores, Wu et al.72 demonstraram que trabalhadores de fornos de coqueria, homozigotos para o genótipo MspI (CYP1A1*2A/*2A), apresentaram diferença estatisticamente significante para os níveis de 1-OHPyr urinário, em relação aos heterozigotos e homozigotos normais. Alexandrie et al.73 associaram os níveis de 1-OHPyr a vários polimorfismos em um grupo exposto a PAHs que trabalhavam em refinaria de alumínio e demonstraram maiores níveis desse metabólito nos indivíduos com os genótipos CYP1A1 Ile/Val e GSTM1 nulo.
Nosso grupo está realizando um estudo para avaliar a influência dos genótipos GSTT1, GSTM1, GSTP1, CYP1A1*2A, CYP1A1*2B e CYP1A1*4 na excreção urinária de 1-hidroxipireno e na atividade mutagênica da urina em cortadores de cana-de-açúcar não fumantes.
De maneira geral, a população como um todo está constantemente exposta às complexas misturas de compostos químicos ambientais que podem causar danos à saúde, expressos a curto ou longo prazo, acentuando-se ainda mais em indivíduos ocupacionalmente expostos, o que mostra a importância do monitoramento dos mesmos. Assim sendo, a correlação de biomarcadores de suscetibilidade e de exposição interna, é bastante eficiente no estabelecimento de estratégias preventivas para tais grupos.
Conclusões
Muitos estudos têm demostrado a importância dos polimorfismos fenotípicos no metabolismo dos compostos químicos como fatores de risco no desenvolvimento tanto do câncer quanto de outras doenças associadas à exposição aos poluentes químicos. Paralelamente a esses estudos, as bases genéticas de muitos dos polimorfismos foram elucidados e métodos não invasivos, como a genotipagem, foram desenvolvidos, com a vantagem de serem aplicados em grandes populações. A identificação de fatores genéticos que, agindo independentemente ou em conjunto com a exposição ambiental, aumenta o risco de toxicidade ou câncer, tem importantes implicações na prevenção, diagnóstico precoce e intervenção as doenças humanas, pois uma vez identificados os grupos suscetíveis, esses podem ser persuadidos a evitar conhecidos fatores de risco. Políticas públicas de redução na emissão de gases resultantes de combustão automotiva (exigência legal de filtros catalíticos em veículos a diesel, gasolina e álcool), obrigatoriedade de filtros eletromagnéticos em chaminés industriais, equipamento de proteção para o trabalhador sujeito à exposição ocupacional podem significar uma importante redução na incidência do câncer na população e nos gastos com saúde pública.
Los autores no manifiestan conflictos.
Bibliografía del artículo
- UNICA. União da AgroindUstria Canavieira de São Paulo. 2004; http://www.Unica.com.br/pages/sociedade_desenvol1.asp.
- UNICA. União da AgroindUstria Canavieira de São Paulo. 2004; http://www.Unica.com.br/files/informacaounica/unica57.pdf.
- Schoket B. DNA Damage in humans exposed to environmental and dietary polycyclic aromatic hydrocarbons. Mutat Res 1999; 424(2):143-53.
- Van Houdt JJ. Mutagenic activity of airborne particulate matter in indoor and outdoor environments. Atmos Environ 1990;24B:207-20.
- Lewtas J, Lewis C, Zweidinger R, et al. Sources of genotoxicity and cancer risk in ambient air. Pharmacogenetics 1992;2(6):288-96.
- Hartzell, GE. Overview of combustion toxicology. Toxicology 1996;115:7-23.
- Deportes I, Guyod J, Zmirou D. Hazard to man and the environment posed by the use of urban waste compost: a review. The Sci of the Total Environ 1995;172:197-222.
- Bartsch H. Studies on biomarkers in cancer etiology and prevention: a summary and challenge of 20 years of interdisciplinary research. Mutat Res 2000;462:255-79.
- EPA. Environmental Protection Agency. Method TO-13A determination of polycyclic aromatic hydrocarbons (PAHs) In ambient air using gas chromatography/mass spectrometry (GC/MS). Washington: Environmental Protection Agency: 1999. p.78.
- NIOSH. National Institute of Occupational Safe and Health. Polynuclear aromatic hydrocarbons by HPLC: Method 5506. Cicinatti: 1998. p9.
- Dipple A, Chaucheng S, Bigger A. Polycycliclic aromatic hydrocarbon carcinogens. Mutag Carcinog Diet 1990:109-27.
- Miller E, Miller J. The mutagenicity of chemical carcinogens: correlations, problems and interpretations . In: Hollaender A, editor. Chemical Mutagen. New York:Plenum; 1971.p.83-119.
- Vo Dinh T. Chemical analysis of polycyclic aromatic compounds. In: Vicent JH, editor. Aerosol samplim. New York:John Wiley and Sons; 1989. p.17-21.
- Kim SJ, Jajoo HK, Kim HY, et al. An efficient route to N6 deoxyadenosine adducts of diol epoxide of csrcinogenic polycyclic aromatic hydrocarbons. Bioorg. Med. Chem 1995;3(6):811-22.
- Peltonen K, Dipple A. Polycyclic aromatic hydrocarbons: chemistry of DNA adduct formation. J Occup Environ Med 1995;37(1):52-8.
- Dipple A, Moschel R. Chemistry of DNA alkylation and aralkylation. In Mutation and Environmental. Wiley:Liss Inc; 1990. p.71-80.
- Doerjer G, Bedell MA, Oesch F. DNA adducts and their biological relevance. In: OBE G, editor. Mutations in Man. Berlin: Springer-Verlag; 1984. p.20-34.
- Canella K, Peltonen K, Dipple A. Identification of (+) and (-) anti benzo(a)pyrene dihydrodiol epoxide-nucleic acid adducts by the 32P-postlabeling assay. Carcinogenesis 1991;12:1826-31.
- Weston A, Bowman E, Manchester D, Harris C. Fluorescence detection of lesions in DNA. In: Sutherland B, Woodheah A, editores. DNA Damage and Repair in Human Tissues. New York:Plenum; 1990. p.63-81.
- IARC. International Agency for Research on Cancer. 2003; http://193.51.164.11/monoeval/crthall.html.
- Sakiara KA. Otimização e validação de metodologia para determinação de 1-hidroxipireno em urina [dissertação]. Araraquara (SP): Instituto de Química/UNESP; 2001.
- WHO. World Health Organization. Selected non-heterocyclic aromatic hydrocarbons. Genova: 1998. p.883.
- Friedberg GC, Walker, GC, Siede, W. DNA repair and mutagenesis. Washington: ASM Press; 1995.
- Barret JC. Mechanisms of multistep carcinogenesis and carcinogen risk assessment. Environ Health Perspect 1993;100:9-20.
- Lewin, B. Genes VI. Oxford: Oxford University Press; 1997.
- Cohen LA. Diet and Cancer. Sci. Am 1987;257:42-8
- Yamasaki H, Asby J, Bignami M, et al. Nongenotoxic carcinogens: development of detection methods based on mechanisms: a European project. Mutat Res 1996;353:47-63.
- Sikora K. Developing a Global Strategy for Cancer. Eur J Cancer 1999;35:1870-7.
- Ramel C. Pollution, carcinogenesis and cancer prevention. Acta Oncol 1991;30:27-33.
- Delclos KB, Manjanatha, MG, Li, EE, et al. 32P- postlabelling in studies of arylamine and nitroatomatic hydrocarbon activation and mutagenesis. IARC Sci Publ 1993;124:79-86.
- Davidson BJ, Hsu TC, Schantz SP. The genetics of tabacco-induced malignancy. Arch Otolaryngol Head Neck Surg 1993;199:1198-205.
- Scully C. Oncogenes, tumor suppressors and viruses in oral squamous carcinoma. J Oral Pathol Med 1993;22:337-47.
- Kaderlik KR, Kadlubar FF. Metabolic polymorphism and carcinogen-DNA adduct formation in human populations. Pharmacogenetics 1995;5:108-17.
- Ames BN, Gold LS, Willett VC. The causes and prevention of cancer. Proc Natl Acad 1995;92:5258-65.
- Motykiewicz G, Michalska J, Pendzich J, et al. A cytogenetic study of men envoronmentally and occupationally exposed to airbone pollutants. Mutat Res 1992;280:253-9.
- Hatjian BA, Edwards JW, Harrison J, et al. Ambient, biological and biological effect monitorng of exposure to polycyclic aromatic hydrocarbons (PAHs). Toxicol Lett 1995;77:271-279.
- Sato MI, Valent GU, Coimbrão CA, et al. Mutagenicity of airborne particulate organic material from urban and industrial areas of Sao Paulo, Brazil. Mutat Res 1995;335:317-30.
- Vargas VMF, Horn, RC, Guidobono RR, et al. Mutagenic activity of. aerborne particulate matter from the urban area of. Porto Alegre, Brazil. Genet Mol Biol 1998;21:247-53.
- Zamperlini GCM, Silva MRS, Vilegas W. Identification of polycyclic hidrocarbons in sugar cane soot by gas chromatography-mass spectrometry. Chromatographia 1997;46:655-63.
- Bosso RMV. Avaliação da atividade mutagênica da fuligem sedimentável proveniente da queima da cana-de açUcar e da urina dos cortadores de cana através de ensaios com mutação gênica reversa em Salmonella typhimurium [dissertação]. São José do Rio Preto (SP): Instituto de Biociências, Letras e Ciências Exatas/Universidade Estadual Paulista; 2000.
- Amre DK, Infante-Rivard C, Dufresne A, et al. Case-Control study of lung cancer among sugar cane farmers in India. Occup Environ Med 1999;56:548-52.
- Pavanello S, Clonfero E. Biological indicadors of genotoxic risk and metabolic polymorphisms. Mutat Res 2000;465:285-308.
- Jongeneelen F. Methods for routine biological monitoring of carcinogenic PAH- mixtures. Science Total Environ 1987;199:141-9.
- Dor F, Dab W, Emperur-Bissonnet P, Zmirou D. Validity of Biomarkers in Environmental Health studies: the case of PAHs and benzene. Crit Rev Toxicol 1999;29(2):129-68.
- Singh R, Tucek M, Maxa K, et al. A rapid and simple method for the analysis if 1-hydroxypyrene glucuronide: a potential biomarker for polycyclic aromatic hydrocarbon exposure. Carcinogenesis 1995;16(12):2909-15.
- Jongeneelen F. Biological Monitoring of environmental exposure to polycyclic aromatic hydrocarbons: 1-hydroxypyrene in urine of people. Toxicol lett 1994;72:205-11.
- Kriek E, Rojas M, Alexandrov K, et al. Polycyclic aromatic hydrocarbon-DNA adducts in humans: relevance as biomarkers for exposure and cancer risk. Mutat Res 1998;400:215-31
- Perera FP, Weinstein IB. Molecular epidemiology: recent advances and future directions. Carcinogenesis 2000;21:517-24.
- Lang M, Pelkonen O. Metabolism of xenobiotics and chemical carcinogenesis. IARC 1999;148:13-22.
- Kawajiri K, Watanabe J, Gotoh O, et al. Structure and drug inducibility of the human cytochrome P-450c gene. Eur J Biochem 1986;159:219-25.
- Taningher M, Malacarne D, Izzotti A, et al. Drug metabolism polymorphisms as modulators of cancer susceptibility. Mutat Res 1999;436:227-61.
- The human cytochrome P450 (cyp) allele nomenclature committee. 2004; http//www.imm.ki.se/CYPalleles.
- Garte S. The role of ethnicity in cancer susceptibility gene polymorphisms: the example of CYP1A1. Carcinogenesis. 1998;19:1329-32.
- Kawajiri K, Nakachi K, Imai K, et al. Identification of genetically high risk individuals to lung cancer by DNA polymorphisms of cytochrome P450 1A1 gene. FEBS Let 1990;263(1):131-3.
- Hayashi S-I, Watanabe J, Nakachi K, et al. PCR detection of na A/G polymorphismwith exon 7 of the CYP1A1 gene. Nucleic Acid Res 1991;19:47-97.
- Hamada GS, Sigimura H, Suzuki I, The heme-binding region polymorphism of cytochrome P4501A1 (CYP1A1), rather than the RstI polymorphism of IIE1 (CYPIIE1), is associated with lung cancer in Rio de Janeiro. Cancer Epidemiol Biomark Prev 1995;4:63-67.
- Wormhoudt LW, Commandeur JNM, Vermeulen NPE. Genetic polymorphisms of human N-acetiltransferase, Cytochrome P450, Glutathione-S-Transferase, and Epoxide Hydrolase Enzymes: relevance to xenobiotic metabolism and toxicity. Crit Rev Toxicol 1999;29:59-124.
- Abdel-Rahman SZ, El-Zein RA, Anwar, WA, et al. A multiplex PCR procedure for polymorphic analysis of GSTM1 and GSTT1 genes in population studies. Cancer Lett 1996;107:229-33.
- Viezzer C, Norppa H, Clonfero E, et al. Influence of GSTM1, GSTT1, GSTP1, and EPHX gene polymorphisms on DNA adduct level and HPRT mutant frequency in coke-oven workers. Mutat Res 1999;431:259-69.
- Abdel-Rahman SZ, Anwar WA, Abdel-Aal WE, et al. GSTM1 and GSTT1 genes are potential risk modifiers for bladder cancer. Cancer Detect Prev 1998;22:129-38.
- Seidegard J, Pero RW, Markowitz MM, et al. Isoenzyme (s) of glutathione transferase (class Mu) as a marker for the susceptibility to lung cancer: a follow-up study. Carcinogenesis 1990;11:33-6.
- Bell DA, Thompson CL, Taylor JA, et al. Genetic monitoring of human polymorphic cancer susceptibility genes by polymerase chain reaction: application to glutathione transferase . Environ Health Perspect 1992;98:113-17.
- Rossit AR, Cabral IR, Hackel CB, et al. Polymorphismsin the DNA repair gene XRCC1 and susceptibility to alcoholic liver cirrhosis in older southeastern Brazilians. Cancer Lett 2002;180:173-82.
- Hirvonen A, Nylund L, Kociba P, et al. Modulation of mutagenicity by genetically determined carcinogen metabolism in smokers. Carcinogenesis 1994;15:813-15.
- Binkovà B, Lewtas J, Mìskova I, et al. Biomarker studies in Northern Bohemia. Environ Health Perspec 1996;104:591-97.
- Gabbani G, Hou SM, Nardini B, et al. GSTM1 and NAT2 genotypes and urinary mutagens in coke oven workers. Carcinogenesis 1996;17:1675-81.
- Gabbani G, Pavanello S, Nardini B, Influence of metabolic genotype GSTM1 on levels of urinary mutagens in patients treated topically with coal tar. Mutat Res 1999;440:27-33.
- Costa DJ, Slott V, Binkova, et al. Influence of GSTM1 and NAT2 genotypes on the relationship between personal exposure to PAHs and biomarkers of internal dose. Biomarkers 1998;3:411-24.
- Hemminki K, Dickey C, Karlsson S, et al. Aromatic DNA adducts in foundry workers in relation to exposure, life style and CYP1A1 and glutatione transferase M1 genotype. Carcinogenesis 1997;18:345-50.
- Merlo F, Andreassen A, Weston A, et al. Urinary excretion of 1-hydroxypyrene as a marker for exposure to urban air levels of polycyclic aromatic hydrocarbons, Cancer Epidemiol. Biomarkers Prev 1998;7:147-55.
- Pavanello S, Gabbani G, Mastrangelo G, et al. Influence of GSTM1 genotypes on anti-BPDE-DNA adduct levels in mononuclear white blood cells of humans exposed to PAHs. Int Arch Occup Environ Health 1999;72:238-46.
- Wu MT, Huang SL, Ho CK, et al. Cytochrome P450 1A1 MspI polymorphism and urinary 1-hydroxypyrene concentrations in coke-oven workers, Cancer epidemiol. Biomarkers Prev 1998;7:823-29.
- Alexandrie AK, Warholm M, Carstensen U, et al. CYP1A1 and GSTM1 polymorphisms affect urinary 1-hydroxypyrene levels after PAH exposure. Carcinogenesis 2000;21:669-76.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC


")
