PESQUISA NEONATAL DE FIBROSIS QUISTICA: EVALUACION DEL PROTOCOLO TIR/ADN (ANALISIS DE LAS MUTACIONES MULTIPLES DEL GEN CFTR)/TIR EN UNA POBLACION DE MAS DE 250 000 NEONATOS
(especial para SIIC © Derechos reservados)
Coautores
Manuela Seia* Carlo Corbetta**
Clinical Research Molecular Genetic Laboratory, A.O. Istituti Clinici di Perfezionamento, Milano*
Neonatal Screening Centre, A.O. Istituti Clinici di Perfezionamento, Milano**
Recepción del artículo: 20 de agosto, 2004
Aprobación: 0 de , 0000
 Conclusión breve
Valoración de un programa de pesquisa neonatal de fibrosis quística aplicado en el período 1998-2001: su eficiencia, sensibilidad y especificidad.
Resumen
Objetivo: Evaluación de un programa de dos etapas para la pesquisa neonatal de fibrosis quística en una cohorte de niños nacidos entre el 1/10/98 y el 31/12/01 en la región de Lombardía, noroeste de Italia. Métodos: El programa de detección se basó en la evaluación de TIR/ensayo de mutaciones múltiples del gen CFTR/TIR. El diagnóstico se confirmó mediante prueba del sudor positiva y por medio de un extenso análisis genético molecular (DGGE) que mostró dos alteraciones del gen CFTR en los niños que presentaron una prueba del sudor con valores en el límite superior de lo normal. Resultados: Se evaluaron 280 354 neonatos; 4 719 lactantes hipertripsinémicos (1.68%) mediante el estudio de un panel de 31 mutaciones en el gen CFTR. Se identificaron 34 neonatos con dos mutaciones en el gen CFTR, se diagnosticaron 32 casos de FQ (fibrosis quística) entre los niños que presentaban una sola mutación, quienes repitieron la prueba del sudor, y se identificaron 5 casos de FQ entre los neonatos con hipertripsinemia persistente. En los niños con valores en el límite superior de lo normal en la prueba del sudor (30-40 mmol/l), se llevó a cabo un extenso análisis genético molecular del gen CFTR y se diagnosticaron más de 14 formas leves de la enfermedad. En este período sólo se diagnosticó FQ en dos niños fuera del programa de pesquisa, y se determinó de esta manera una incidencia global de FQ de 1:3 222. Conclusiones: El protocolo TIR/ADN (ensayo de las mutaciones múltiples del gen CFTR)/TIR permite realizar un diagnóstico temprano de la forma clásica de FQ. En recién nacidos hipertripsinémicos heterocigotas la confirmación o exclusión de FQ puede ser dificultosa. Se recomienda un amplio análisis genético molecular para estos niños y la consideración de nuevos valores normales de cloruro en el sudor para los primeros meses de vida.
Palabras clave
Fibrosis quística, CFTR, pesquisa neonatal, fibrosis quística atípica, prueba del sudor con valores en el límite sup
Clasificación en siicsalud
Conclusión breve
Valoración de un programa de pesquisa neonatal de fibrosis quística aplicado en el período 1998-2001: su eficiencia, sensibilidad y especificidad.
Resumen
Objetivo: Evaluación de un programa de dos etapas para la pesquisa neonatal de fibrosis quística en una cohorte de niños nacidos entre el 1/10/98 y el 31/12/01 en la región de Lombardía, noroeste de Italia. Métodos: El programa de detección se basó en la evaluación de TIR/ensayo de mutaciones múltiples del gen CFTR/TIR. El diagnóstico se confirmó mediante prueba del sudor positiva y por medio de un extenso análisis genético molecular (DGGE) que mostró dos alteraciones del gen CFTR en los niños que presentaron una prueba del sudor con valores en el límite superior de lo normal. Resultados: Se evaluaron 280 354 neonatos; 4 719 lactantes hipertripsinémicos (1.68%) mediante el estudio de un panel de 31 mutaciones en el gen CFTR. Se identificaron 34 neonatos con dos mutaciones en el gen CFTR, se diagnosticaron 32 casos de FQ (fibrosis quística) entre los niños que presentaban una sola mutación, quienes repitieron la prueba del sudor, y se identificaron 5 casos de FQ entre los neonatos con hipertripsinemia persistente. En los niños con valores en el límite superior de lo normal en la prueba del sudor (30-40 mmol/l), se llevó a cabo un extenso análisis genético molecular del gen CFTR y se diagnosticaron más de 14 formas leves de la enfermedad. En este período sólo se diagnosticó FQ en dos niños fuera del programa de pesquisa, y se determinó de esta manera una incidencia global de FQ de 1:3 222. Conclusiones: El protocolo TIR/ADN (ensayo de las mutaciones múltiples del gen CFTR)/TIR permite realizar un diagnóstico temprano de la forma clásica de FQ. En recién nacidos hipertripsinémicos heterocigotas la confirmación o exclusión de FQ puede ser dificultosa. Se recomienda un amplio análisis genético molecular para estos niños y la consideración de nuevos valores normales de cloruro en el sudor para los primeros meses de vida.
Palabras clave
Fibrosis quística, CFTR, pesquisa neonatal, fibrosis quística atípica, prueba del sudor con valores en el límite sup
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/69865
Especialidades
 Principal: Diagnóstico por Laboratorio, Pediatría,
Principal: Diagnóstico por Laboratorio, Pediatría,
Relacionadas: Bioquímica, Endocrinología y Metabolismo, Medicina Interna, Neumonología,
Enviar correspondencia a:
Rita Padoan. Cystic fibrosis Service. Ospedale dei Bambini. Piazzale Spedali Civili 1. 25123 Brescia, Italia Padoan, Rita
Patrocinio y reconocimiento
Agradecimiento. Agradecemos a la profesora Anna Bossi (del Instituto de Estadísticas Médicas y Biometría de la Universidad de Milán) por la información del Registro Italiano de FQ y los análisis estadísticos; a Tiziana Mariani, Cinzia Coltellaro, E. Manzoni y A. Di Modugno (técnicas), del Centro de Pesquisa Neonatal, y a Antonella Ambrosioni, Sabrina Fiori y G. Porcaro (biólogas), del Laboratorio de Genética Molecular, por su asistencia.
NEWBORN SCREENING FOR CYSTIC FIBROSIS: EVALUATION OF AN IRT/DNA (MULTIPLE-CFTR-MUTATIONS ANALYSIS)/IRT PROTOCOL IN A POPULATION OF OVER 250 000 NEWBORNS
Abstract
Objective: To evaluate a two-tier neonatal screening program for cystic fibrosis (CF), on a cohort of newborns born in the period 1.10.98-31.12.01 in Regione Lombardia, North-western Italy. Methods: The screening program was based on IRT/multiple CFTR mutations assay/IRT assessment. Diagnosis was confirmed by means of a positive sweat test and/or by means of a wide molecular genetic analysis (DGGE) showing two alterations of the CFTR gene in infants presenting with a borderline sweat test. Results: 280 354 newborns were screened. 4 719 hypertrypsinaemic infants (1.68%) were analysed using a panel of 31 CFTR mutations. 34 neonates with two CFTR mutations were identified. 32 CF were diagnosed among infants presenting only one mutation, recalled for sweat test. 5 CF were identified among persistent hypertrypsinaemic newborns. A wide genetic analysis of the CFTR gene was performed in infants with borderline sweat test (30-40 mmol/l), diagnosing further 14 mild forms of the disease In this period only two children were diagnosed as CF apart of the screening program, thus leading to an overall CF incidence of 1:3 222. Conclusions: IRT/DNA(multiple CFTR mutations assay)/IRT protocol results in an early diagnosis of the classic form of CF. In heterozygotes hypertrypsinaemic newborns confirmation or exclusion of CF may be difficult. A wide genetic molecular analysis is recommended for these infants and a new value for normal sweat chloride in the first months of life should be considered.
Key words
Cystic fibrosis, CFTR, newborn screening, atypical cystic fibrosis, borderline sweat test
PESQUISA NEONATAL DE FIBROSIS QUISTICA: EVALUACION DEL PROTOCOLO TIR/ADN (ANALISIS DE LAS MUTACIONES MULTIPLES DEL GEN CFTR)/TIR EN UNA POBLACION DE MAS DE 250 000 NEONATOS
(especial para SIIC © Derechos reservados)
Artículo completo
Introducción
La fibrosis quística (FQ) es el trastorno autosómico recesivo más frecuente entre las personas de raza blanca, con una frecuencia en Italia de 1:4 231 nacidos vivos (datos del Registro Nacional).1 Las mutaciones en el gen regulador de la conducción transmembrana de la fibrosis quística (CFTR), localizado en el cromosoma 7, producen FQ.2,3 El gen codifica para una proteína que se expresa en la membrana apical de determinadas células epiteliales. El CFTR funciona principalmente como un canal de cloro regulado por AMPc.4 Desde que comenzó la clonación de genes se identificaron más de 1 000 mutaciones (que fueron notificadas al CF Genetics Consortium)5 con una gran variabilidad en los fenotipos.6 Además de la mutación delF508, responsable en gran proporción de los alelos mutados en la mayoría de las poblaciones, hay otras 15 a 20 mutaciones que comprenden del 2% al 15% de los alelos de la fibrosis quística, y se comprobó una gran variabilidad genética entre poblaciones.7 Esta enfermedad, potencialmente mortal, se distingue por una o más características de gravedad variable, tales como disminución progresiva de la función pulmonar secundaria a infecciones pulmonares crónicas, insuficiencia exocrina pancreática que determina desnutrición y retraso en el crecimiento, enfermedad hepática y disminución de la reabsorción de los iones cloruro del sudor.
A pesar de la relativa frecuencia de FQ en las poblaciones de raza blanca y los síntomas claros de la forma clásica de la enfermedad, puede producirse una demora en el diagnóstico que determine desnutrición grave o enfermedad pulmonar crónica en los primeros años de vida. Para realizar el diagnóstico y el tratamiento tempranos se implementaron programas de pesquisa neonatal desde comienzos de la década del ’80, con diferentes estrategias en distintos países.8,9 Actualmente, se considera que la hipertripsinemia neonatal sumada a un análisis del gen CFTR mediante un gran panel de mutaciones10 constituye un buen método de pesquisa: sus ventajas son tanto su alta sensibilidad como su alto poder predictivo, lo cual junto con el diagnóstico temprano de FQ dentro del primer mes de vida, permite un mejor enfoque clínico. La derivación temprana para su tratamiento a un centro especializado11 determina menor morbilidad, beneficios nutricionales, mejores funciones cognitivas y mayor expectativa de vida, así como el oportuno asesoramiento genético para la familia.12-16
Una consecuencia controvertida de los procedimientos genético-moleculares aplicados en los programas de pesquisa neonatal es la detección de portadores para las mutaciones del gen CFTR y la identificación de un gran número de niños con FQ heterocigotas detectados en el período neonatal, que constituye un gran desafío desde el punto de vista ético. En este grupo de pacientes puede ser difícil la confirmación o la exclusión de FQ, y casi siempre demanda tiempo; por ende, la dificultad diagnóstica puede provocar ansiedad, malentendidos y estrés familiar.
El programa de pesquisa neonatal para FQ se implementó en 1983 en la región de Lombardía (al noroeste de Italia), por medio de las determinaciones de tripsina inmunorreactiva (TIR-b) en las tarjetas de Guthrie, como un tercer programa adicional dentro del panel preexistente para fenilcetonuria (PKU) e hipotiroidismo congénito. El programa de detección se revisó y perfeccionó con las estrategias subsecuentes, agregándose el análisis molecular (búsqueda de la mutación delF508) en 1993 a las determinaciones de TIR-b. La evaluación de estas estrategias (TIR aislado y TIR más la búsqueda de la mutación delF508) mostró un gran número de falsos negativos en el programa de pesquisa debido principalmente a la gran heterogeneidad genética de la FQ en la población italiana.17 Desde octubre de 1998 se agregó la búsqueda de 31 mutaciones del gen CFTR al programa de pesquisa (TIR/ADN/TIR) en un esfuerzo por mejorar su eficiencia. Además, y al igual que en nuestra experiencia previa, algunos niños fueron correctamente seleccionados por su hipertripsinemia neonatal persistente pero recibieron un diagnóstico tardío de FQ debido al resultado de una prueba del sudor que se consideró normal (Cl- < 60 mM/l) en los primeros meses de vida.18 A los niños que presentaron una prueba del sudor poco definitoria en los primeros tres meses de vida se les ofreció un amplio análisis genético del gen CFTR y un seguimiento clínico para excluir o confirmar el diagnóstico de FQ.
El objetivo del presente estudio fue evaluar nuestro programa de pesquisa en el período 1998-2001, su eficiencia, sensibilidad y especificidad, junto con los nuevos desafíos que presenta el análisis molecular: la detección de un gran número de portadores entre los recién nacidos hipertripsinémicos y las dificultades diagnósticas en presencia de una prueba del sudor con valores en el límite superior de lo normal.
Materiales y métodos
En el período comprendido entre el 1 de octubre de 1998 y el 31 de diciembre de 2001 se evaluaron 280 354 recién nacidos mediante la estrategia en dos etapas TIR/ADN (ensayo de mutaciones múltiples del gen CFTR - panel de 31 mutaciones)/TIR, como se informó previamente.19 La figura 1 muestra los pasos del procedimiento de pesquisa. Los recién nacidos con dos mutaciones en el gen CFTR fueron derivados directamente al centro de FQ, la prueba del sudor por iontoforesis con pilocarpina20 se llevó a cabo inmediatamente en niños con una mutación detectada en el gen CFTR o con una segunda determinación de TIR por encima del valor de corte. Se consideró patológica una prueba del sudor con valores de cloruro superiores a 40 mmol/l.21 Un valor de cloruro en la prueba del sudor mayor de 30 mmol/l se considera en el límite y requiere mayores investigaciones.

En lactantes que presentan prueba del sudor positiva o en el límite, en los cuales ninguno o sólo un alelo CFTR fue identificado, se llevó a cabo un amplio análisis molecular por la técnica de electroforesis en gel de gradiente desnaturalizante (DGGE), como fuera previamente descrito.22 Se utilizó un estudio por PCR específico para alelos para distinguir los alelos 5T, 7T y 9T. Los productos de amplificación se visualizaron por una tinción en gel de agarosa con etilbromuro al 4%.
Se llevó a cabo una revisión de la base de datos de los pacientes con diagnóstico de FQ seguidos en el Centro de FQ de Milán, nacidos en el período 1998-2001, con resultados negativos en el programa de detección neonatal. La misma información se solicitó al Registro Italiano de FQ, con respecto a los pacientes nacidos en esta región y seguidos en diferentes centros de FQ.
Resultados
En la región de Lombardía, en el período comprendido entre el 1 de octubre de 1998 y el 31 de diciembre de 2001, la tasa de cobertura del programa de pesquisa neonatal fue mayor al 99% de todos los nacimientos, y se evaluaron 280 354 neonatos. Se seleccionaron 4 719 recién nacidos hipertripsinémicos (1.68%) para el análisis de mutaciones, mediante un ensayo para mutaciones múltiples del gen CFTR.
Se detectó íleo meconial en 11 niños, todos con resultados positivos en el programa de pesquisa.
Entre los pacientes hipertripsinémicos, 34 eran homocigotas o heterocigotas compuestos para las mutaciones del gen CFTR consideradas en el panel, por lo cual recibieron diagnóstico de FQ y derivación temprana a la clínica de FQ en el primer mes de vida.
Entre los individuos heterocigotas, se diagnosticó FQ en 32 niños con niveles de cloruro en el sudor > 40 mmol/l en forma persistente, por lo cual se derivaron al Centro de FQ para su evaluación clínica; mientras que los niños con valores de cloruro en el sudor en el límite de lo normal (> 30 mmol/l a < 40mM/l) fueron derivados para análisis genético extenso con el fin de confirmar o excluir FQ. Entre ellos, se diagnosticaron 14 pacientes con formas atípicas de la enfermedad.
Los niños con hipertripsinemia sin mutaciones en el gen CFTR fueron sometidos a una segunda toma de muestra de TIR-b a los 20 a 30 días de vida; los niños con una segunda prueba por fuera del valor de corte fueron derivados para una prueba del sudor, y así se confirmó el diagnóstico de FQ en 5 niños.
La FQ se diagnosticó en un total de 85 pacientes en el período de 39 meses: 34 homocigotas o heterocigotas compuestos, 46 niños que presentaron al menos una mutación CFTR identificada por el panel de pesquisa y 5 pacientes que no presentaron mutaciones. Con relación al problema de los resultados negativos del programa de pesquisa, sólo a dos niños nacidos durante el período de observación se les realizó el diagnóstico de FQ leve por los síntomas: al primero, a los 20 meses por una alcalosis metabólica grave con hipocloremia durante el caluroso verano de 2003; mientras que al segundo, a los 3 años, por infecciones recurrentes de la vía aérea superior.
La incidencia global de FQ en esta región fue de 1:3 222 (CI 95% 1:2 653-1:4 102).
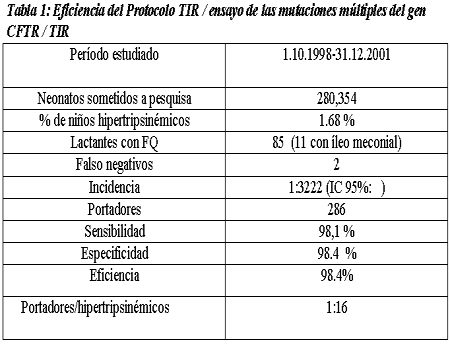
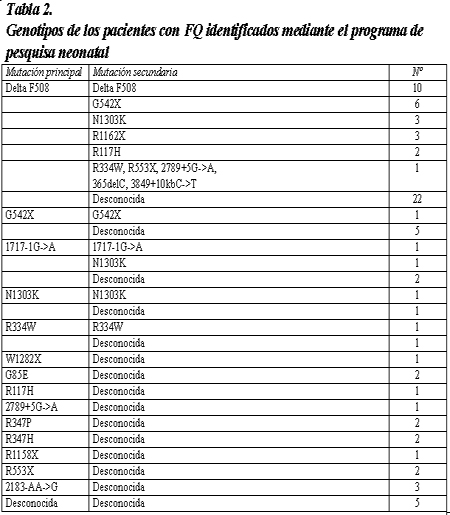
La tabla 1 muestra los resultados de la estrategia TIR/ADN (31 mutaciones)/TIR, en el período considerado. Los genotipos de los pacientes con FQ seleccionados por la pesquisa neonatal se muestran en la tabla 2: 16.6% son homocigotas, 24% heterocigotas compuestos y 54% heterocigotas. Así, alrededor del 95% de los pacientes con FQ presentan al menos un alelo CFTR que fue identificado por el ensayo de PCR/OLA (ensayo de ligadura de oligonucleótidos combinado con PCR). A los niños con FQ (con formas clásicas o atípicas de la enfermedad) que presentaban sólo una mutación o ninguna, se les realizó un análisis genético extendido por la técnica de DGGE y secuenciamiento, y las mutaciones en el gen CFTR identificadas se muestran en la tabla 3.
Además, con la presente estrategia, se identificó en forma efectiva un alto número de portadores: 286 niños entre los recién nacidos hipertripsinémicos (1/16).

Discusión
El programa de pesquisa neonatal de FQ en recién nacidos se implementó a partir de un protocolo previo durante el período 1998-2001 y se elaboró en vista de la alta heterogeneidad genética de esta población.23 La estrategia de detección se basó en un protocolo TIR/ADN (ensayo para mutaciones múltiples del gen CFTR)/TIR, por lo cual el análisis molecular se extendió sustancialmente, con una cobertura cercana al 75% de los alelos mutados en esta población con FQ. Los resultados muestran que casi la totalidad de los niños afectados por la forma clásica de la FQ nacidos en nuestra región se derivan correctamente y en tiempo (antes de las 4 semanas de edad) a un centro de FQ. Además, este protocolo permitió reconocer también formas leves o atípicas de la enfermedad.
Los datos mostraron buenos resultados en términos de sensibilidad (98.1%), especificidad (98.4%) y eficacia (98.4%). Hasta el momento, sólo se diagnosticaron dos casos de FQ atípica, con resultados falsos negativos para el programa de pesquisa. De cualquier forma, la incidencia de FQ en el período examinado fue de 1:3 222, más elevada que la informada para el total de la población italiana.
La frecuencia observada de portadores fue de 1/16 en los recién nacidos hipertripsinémicos, mayor que el valor estimativo de 1/29. Estos datos confirman los hallazgos previos24 acerca de una frecuencia aumentada de heterocigotas para mutaciones en el gen CFTR en neonatos con TIR positiva. En recién nacidos heterocigotas, la confirmación o exclusión de FQ se basa en la prueba de cloruro en el sudor; sin embargo, cierto número de pacientes mostró concentraciones elevadas de cloruro en el sudor, por encima del límite superior de lo normal. Los niveles anormales de cloruro en el sudor deben considerarse una manifestación fenotípica subclínica de FQ en los neonatos hipertripsinémicos portadores de una mutación para FQ. Además, cierto número de estos niños podría ser portador en el otro cromosoma de una segunda mutación leve en el gen CFTR,25 y es bien sabido que los heterocigotas compuestos con alelos CFTR de gravedad y con infrecuentes mutaciones leves podrían no presentar una prueba de cloruro en el sudor diagnóstica al comienzo de su vida.18
De esta forma, se requiere perfeccionar el seguimiento diagnóstico de los neonatos con alteraciones bioquímicas (TIR-b y prueba del sudor) y moleculares (una mutación FQ), detectados por el programa de pesquisa de recién nacidos.
De acuerdo con nuestra experiencia podría lograrse mediante lo siguiente:
a) Centralizar la prueba del sudor que debe llevarse a cabo por personal altamente entrenado y experimentado, con establecimiento de los valores mínimos de referencia para lactantes. Para mejorar la eficacia del programa de pesquisa y para evitar un retraso diagnóstico en niños que presentan mutaciones raras, se modificó el valor superior normal de cloruros para el diagnóstico de FQ, descendiéndolo a 30 mmol/l. Los niños con valores superiores necesitan ser evaluados cuidadosamente en el Centro de FQ.
b) Un amplio estudio genético de la región codificadora para identificar una segunda mutación posible, principalmente en una población con alta heterogeneidad alélica como la italiana, donde la prueba de PCR/OLA es útil solamente para el 75% de los cromosomas FQ.
c) Una elevada colaboración entre el laboratorio y el centro clínico de FQ para el análisis de los casos atípicos. Es necesaria la evaluación completa de los portadores de FQ dentro del programa de pesquisa neonatal para FQ y el centro especializado en FQ debe hacerse cargo de estos pacientes, con visitas clínicas y asesoramiento genético ofrecidos por el equipo médico especializado. Deben llevarse a cabo investigaciones de laboratorio en búsqueda de malabsorción (elastasa fecal y esteatocrito) y Rx de tórax y evaluación microbiológica de las secreciones bronquiales en caso de síntomas respiratorios. Una estricta colaboración con los médicos de atención primaria es también fundamental en estos casos para evitar la información confusa. Este enfoque podría reducir el estrés de los padres, ofrecer un correcto asesoramiento genético y permitir, de ser necesario, el mejor tratamiento en forma temprana.
En conclusión, diversos factores nos llevaron a la obtención de buenos resultados: la eficacia del ensayo molecular aplicado al programa de pesquisa para identificar pacientes de alto riesgo, la mejoría en la seguridad analítica de la detección de TIR-b y en la realización de la prueba del sudor y una nueva definición de los valores normales de laboratorio. Asimismo, dado que la detección neonatal de FQ es un proceso complejo, la cooperación estrecha entre el centro de detección neonatal de FQ (personal del laboratorio de pesquisa, biólogos moleculares) y el centro de FQ clínica (pediatras) permite una derivación temprana de los niños seleccionados por el programa de pesquisa.
Más allá de los resultados, el monitoreo de este proceso requiere ser adecuadamente esquematizado para poder mejorar su rendimiento.
Los autores no manifiestan conflictos.
Bibliografía del artículo
- Bossi A, Casazza G, Padoan R, et al What is the incidence of cystic fibrosis in Italy Data from the National Registry Human Biology 2004, 76(3):455-67.
- Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA Science 1989,245:1066-73.
- Kerem B, Rommens JM, Buchanan JA, et al Identification of the cystic fibrosis gene: Genetic analysis. Science 1989;245:1073-80.
- Welsh MJ, Smith AE Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993 Jul 2;73(7):1251-4.
- cystic fibrosis Genetic Analysis Consortium www.genet.sickkids.on.ca/cftr.
- Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration 2000;67:117-33.
- Bobadilla JL, Macek M, Fine JP et al. 2002. Cystic fibrosis: A worldwide analysis of CFTR mutations: Correlation with incidence data and application to screening. Hum. Mutation19:575–606.
- Crossley JR, Elliott RB, Smith PA Dried blood spot screening for cystic fibrosis in the newborn. Lancet 1979;1:472-74.
- Wilcken D, Wiley V, Sherry G et al. Neonatal screening for cystic fibrosis. A comparison of two strategies for case detection in 1.2 million babies. J Pediatr 1995;127:965-70.
- Wilcken B, Travert G. Neonatal screening for cystic fibrosis: present and future. Acta Paediatr Suppl. 1999 Dec;88(432):33-5.
- Littlewood JM. Neonatal cystic fibrosis screening: a personal overview from the perspective of a pediatrician. In Neonatal screening for cystic fibrosis. Proceedings of the international conference Caen, September 9-11 1998. 1999 Presses Universitaires de Caen, pp309-24.
- Wilcken B, Chalmers G. Reduced morbidity in patients with cystic fibrosis detected by neonatal screening Lancet 1985 Dec 14;2(8468):1319-21.
- Farrell PM, Kosorok MR, Laxova A, et al. Nutritional benefits of neonatal screening for cystic fibrosis. Wisconsin cystic fibrosis Neonatal Screening Study Group. N Engl J Med. 1997 Oct 2;337(14):963-9.
- Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Pediatrics 2001;107:1-13.
- Koscik RL, Farrell PM, Kosorok MR et al. Cognitive function of children with cystic Fibrosis: deleterious effect of early malnutrition Pediatrics 2004;113(6):1549-58.
- Lai HC, Cheng Y, Cho H et al. Association between initial disease presentation, lung disease outcomes, and survival in patients with cystic fibrosis Am J Epidem 2004;159(6):537-46.
- Padoan R, Genoni S, Moretti E, et al. Genetic and clinical features of infants false negative to neonatal screening program for cystic fibrosis Acta Paediatr 2002;91:82-7.
- Padoan R, Bassotti A, Seia M, et al. Negative sweat test in hypertrypsinaemic infants with cystic fibrosis crrying rre CFTR mutations Eur J Pediatr 2002;161:212-5.
- Corbetta C, Seia M, Bassotti A et al. Screening for cystic fibrosis in newborn infants: results of a pilot program based on a two tier protocol (IRT/DNA/IRT) in the Italian population J Med Screen 2002,9:60-3.
- Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilising pilocarpine by iontophoresis. Pediatrics 1959;23:158-61.
- Farrell P, Koscik R. Sweat chloride concentration in infants homozygous or heterozygous for F508 cystic fibrosis. Pediatrics Vol.97 No.4 April 1996.
- Cremonesi L, Ferrari M, Belloni E, Magnani C, Seia M, Ronchetto P et al. Four new mutations of the CFTR gene (541delC, R347H, R352Q, E585X) detected by DGGE analysis in Italian CF patients, associated with different clinical phenotypes. Hum Mutat. 1992;1(4):314-9.
- Rendine S, Calafell F, Cappello N, et al. Genetic history of cystic fibrosis mutations in Italy. I. Regional distribution. Ann Hum Genet. 1997 Sep;61 ( Pt 5):411-24.
- Laroche G, Travert G. Abnormal frequency of delF508 mutation in neonatal transitory hypertrypsinaemia. Lancet 1991;337:55.
- Boyne J, Evans S, Pollitt RJ, et al. Many delF508 heterozygote neonates with transient hypertrypsinaemia have aa second, mild CFTR mutation J Med Genet 2000 ;37(7):543-47.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC


