ENFOQUE CLINICO Y DE LABORATORIO DEL AUMENTO DE LA FUNCION Y NUMERO DE PLAQUETAS
(especial para SIIC © Derechos reservados)
Coautores
Yu-Min Shen* Ravindra Sarode** Eberhard Mammen*** Eugene Frenkel****
MD, Assistant Professor, Internal Medicine. Department of Medicine, The University of Texas Southwestern Medical Center at Dallas, Dallas, Texas*
MD, Associate Professor of Pathology. Department of Pathology, The University of Texas Southwestern Medical Center at Dallas, Dallas, Texas**
MD, Professor of Obstetrics and Gynecology. Department of Obstetrics and Gynecology, Wayne State University School of Medicine, Detroit, MI***
MD, Professor, Internal Medicine. Department of Medicine, The University of Texas Southwestern Medical Center at Dallas, Dallas, Texas****
Recepción del artículo: 4 de noviembre, 2004
Aprobación: 0 de , 0000
 Conclusión breve
La trombosis es la manifestación clínica principal del síndrome de hiperactividad de las plaquetas y de la trombocitemia esencial. La heterogeneidad de los sitios afectados por la trombosis es una de las características que sugieren estos cuadros.
Resumen
Las plaquetas desempeñan un papel crítico en la hemostasia, de manera tal que las alteraciones de la función y del número de plaquetas pueden desencadenar estados trombohemorrágicos. Está demostrado que el aumento de la reactividad plaquetaria (plaquetas hiperactivas) se observa en una variedad de estados clínicos o genéticos que pueden concluir en acontecimientos trombóticos. Además, el aumento de la función de las plaquetas puede coexistir con alteraciones en el número de éstas en situaciones de trombocitosis o trombocitemia. En este trabajo se realiza una revisión de los aspectos clínicos, fisiopatológicos y de laboratorio relacionados con el síndrome de hiperactividad plaquetaria. También se describe cómo reconocer los aspectos clínicos y el enfoque diagnóstico y terapéutico de la trombocitosis benigna con aumento en el número de plaquetas y de la lesión mieloproliferativa de la trombocitemia.
Palabras clave
Síndrome de hiperactividad de plaquetas, trombocitosis, trombocitemia
Clasificación en siicsalud
Conclusión breve
La trombosis es la manifestación clínica principal del síndrome de hiperactividad de las plaquetas y de la trombocitemia esencial. La heterogeneidad de los sitios afectados por la trombosis es una de las características que sugieren estos cuadros.
Resumen
Las plaquetas desempeñan un papel crítico en la hemostasia, de manera tal que las alteraciones de la función y del número de plaquetas pueden desencadenar estados trombohemorrágicos. Está demostrado que el aumento de la reactividad plaquetaria (plaquetas hiperactivas) se observa en una variedad de estados clínicos o genéticos que pueden concluir en acontecimientos trombóticos. Además, el aumento de la función de las plaquetas puede coexistir con alteraciones en el número de éstas en situaciones de trombocitosis o trombocitemia. En este trabajo se realiza una revisión de los aspectos clínicos, fisiopatológicos y de laboratorio relacionados con el síndrome de hiperactividad plaquetaria. También se describe cómo reconocer los aspectos clínicos y el enfoque diagnóstico y terapéutico de la trombocitosis benigna con aumento en el número de plaquetas y de la lesión mieloproliferativa de la trombocitemia.
Palabras clave
Síndrome de hiperactividad de plaquetas, trombocitosis, trombocitemia
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/70850
Especialidades
 Principal: Hematología,
Principal: Hematología,
Relacionadas: Bioquímica, Diagnóstico por Laboratorio, Medicina Interna,
Enviar correspondencia a:
Eugene P. Frenkel, MD. Department of Medicine, The University of Texas Southwestern Medical Center at Dallas, 5323 Harry Hines Blvd., Dallas, Texas 75390-8852, EE.UU. Frenkel, Eugene P
Patrocinio y reconocimiento
Subvencionado por el Raymond D. Nasher Research Program.
CLINICAL AND LABORATORY APPROACH TO PLATELET HYPERFUNCTION AND INCREASED PLATELET NUMBER
Abstract
Platelets play a critical role in hemostasis, thus alterations of platelet function and number may result in clinical thrombohemorrhagic states. It has become clear that increased platelet reactivity (i.e. hyperactive platelets) can be observed in a variety of genetic and clinical states with resultant thrombotic events. In addition, platelet hyperfunction may co-exist with increased platelet number in the circumstances of thrombocytosis and thrombocythemia. Herein we review the clinical, pathophysiological and laboratory issues related to hyperactive platelet syndrome. In addition, clinical recognition, diagnostic and therapeutic approach to increased platelet number in the benign state of thrombocythosis and the myeloproliferative lesion of thrombocythemia are reviewed.
Key words
Hyperactive platelet syndrome, thrombocythosis, thrombocythemia
ENFOQUE CLINICO Y DE LABORATORIO DEL AUMENTO DE LA FUNCION Y NUMERO DE PLAQUETAS
(especial para SIIC © Derechos reservados)
Artículo completo
Introducción
Las plaquetas desempeñan un papel importante en la hemostasia. La adhesión plaquetaria con la activación subsiguiente y agregación en los sitios vasculares dañados lleva a la formación del trombo primario que constituye la principal línea de defensa contra la hemorragia. No obstante, las plaquetas no participan exclusivamente en la hemostasia primaria debido a que también actúan en la hemostasia secundaria, ya que proveen la superficie lipídica procoagulante para los factores de coagulación. La trombina, que es el producto final de la cascada de coagulación, es un potente activador plaquetario; por esto, ambos procesos son mutuamente dependientes.1
En los trastornos hemorrágicos se ha estudiado extensamente el papel de las alteraciones en la función de las plaquetas.2-4 Pero se ha prestado menor atención a la identificación de las circunstancias que cursan con el aumento de la función (plaquetas hiperactivas) así como a los problemas que se asocian con el aumento del número de plaquetas (trombocitosis y trombocitemia), eventos que frecuentemente coexisten. Esta revisión se centra en los aspectos terapéuticos y de laboratorio del incremento en la función y en el número de las plaquetas circulantes.
El concepto de las plaquetas hiperactivas (“adherentes”)
Eberhard Mammen fue el primero en describir el síndrome de plaquetas hiperactivas (“adherentes”), en 1982, y descubrió la relación existente entre la trombosis clínica y el aumento in vitro de la agregación plaquetaria. Este investigador reconoció este síndrome en una mujer de 24 años, embarazada de 7 meses, con infarto agudo de miocardio (IAM). La angiografía coronaria de esta paciente no demostró la presencia de lesiones ateroescleróticas y las pruebas de trombofilia resultaron negativas; sin embargo, a través de los estudios de la función de las plaquetas se observó una respuesta agregante elevada ante la epinefrina y la adenosina difosfato (ADP) en el plasma enriquecido con plaquetas (PRP), que se volvió más pronunciada con la dilución de los agonistas. La historia familiar de esta mujer también era importante. La madre de esta paciente tuvo un IAM cuando estaba embarazada y el hermano de 18 años había sufrido dolor precordial recurrente sin que se hubiese podido identificar una enfermedad cardíaca isquémica. La prueba de trombofilia fue negativa en todos los casos. Resultó interesante que el perfil de hiperagregación plaquetaria en el PRP fue idéntico en los tres casos. Además en la microscopia electrónica –al entrar en contacto con la superficie– la adhesión y agregación de las plaquetas era mayor.
El tratamiento con aspirina en dosis de 81 mg inhibió el aumento de la función de las plaquetas en esta paciente y en el resto de su familia y, al ser interrumpido, reaparecieron las alteraciones en la agregación.5,6 Sobre la base de los hallazgos clínicos y de laboratorio, Mammen definió el SPH como un trastorno autosómico dominante en las plaquetas, que se caracteriza por la presencia de trombosis arterial o venosa secundaria al aumento en la agregación plaquetaria. Sobre la base del aumento de la agregación se identificaron tres tipos de este síndrome, inducido por el ADP y la epinefrina, por epinefrina sola y por ADP solo, con el método óptico de agregación plaquetaria (tipo I, tipo II y tipo III, respectivamente).5,6
Los estudios de Mammen sobre SHP incluían pacientes con IAM o angina de pecho con angiografía normales de las arterias coronarias,7 menores de 45 años, con antecedentes de accidente cerebrovascular trombótico o episodios isquémicos transitorios sin factores de riesgo identificables8 o personas que padecían neuropatía isquémica idiopática y sufrían de ceguera permanente o transitoria.9 En estos estudios se comparó sexo, edad y origen étnico de voluntarios con los de estos pacientes y se demostró que, en estos últimos, la respuesta agregante inducida por el ADP y la epinefrina era significativamente mayor. Además, en la microscopia electrónica se constató un aumento en la adherencia y agregación plaquetarias al entrar en contacto con la superficie. Estos resultados fueron confirmados por otros investigadores en pacientes con eventos trombóticos de origen desconocido.8,10,11 Recientemente, otros trabajos demostraron que, in vitro, la hiperreactividad plaquetaria puede ser inducida por sustancias diferentes del ADP, epinefrina o agonistas.12 Es por esto que el nuevo enfoque para estudiar el tema del aumento de la agregación incluye la estimulación de las plaquetas con una variedad de agonistas en diferentes concentraciones, en sangre entera y en condiciones fisiológicas.
Últimamente, el término “síndrome de hiperactividad de las plaquetas” reemplazó al primero o “síndrome de plaquetas adherentes”, debido a que define mejor el aumento de la función de las plaquetas in vitro y su relación con los acontecimientos trombóticos in vivo.
El SPH aparenta ser un fenómeno complejo, en el que la base genética desempeña un papel importante. No obstante, no se han podido caracterizar completamente muchos de los mecanismos subyacentes relacionados con el aumento de la función plaquetaria, para lo cual se requieren nuevas investigaciones.
La base molecular de la hiperactividad de las plaquetas
Algunas observaciones recientes han aportado datos acerca de los cambios moleculares que alteran la función de las plaquetas y que crean la base para la hiperactividad.
El polimorfismo de las glucoproteínas plaquetarias
Los polimorfismos de las glucoproteínas plaquetarias aparentan ser factores moleculares importantes que pueden influir en la reactividad de estas células. Se ha comunicado que el polimorfismo PlA2 (Pro33Leu) de la glucoproteína IIIa, la cadena β3 del receptor de fibrinógeno podría cambiar la respuesta de las plaquetas a los agonistas con un umbral bajo de activación.13 El ADP y la epinefrina inducen el aumento en la agregación de las plaquetas en las que se detecta PlA2,14 se observa un aumento de la expresión de la selectina P,13 y se realza la unión con el fibrinógeno.15 Algunos estudios demostraron el riesgo aumentado de infarto de miocardio y de sucesos coronarios tempranos en los individuos portadores del alelo PlA2;16,17 sin embargo, otros no han podido confirmar estos hallazgos.18
El polimorfismo C807T de la integrina Ia/IIa, un receptor del colágeno, incrementa la densidad de receptores en la superficie de las plaquetas;19 por este motivo se refuerza la unión con el colágeno y los portadores del alelo 807T podrían presentar hiperactividad de las plaquetas. El polimorfismo C807T ha sido asociado con riesgo aumentado de infarto de miocardio y de accidente cerebrovascular en algunos ensayos clínicos,20,21 pero no en todos.22,23
Diferentes polimorfismos (Met 145, Kozak y un número variable de repeticiones en tándem [VNTR]) de la glucoproteína Ib –un receptor para el factor de Von Willebrand (FvW)– fueron identificados como factores de riesgo de los eventos cardiovasculares y cerebrovasculares isquémicos.24-26 El aumento en la reactividad plaquetaria podría estar relacionado con el siguiente fenómeno a nivel de las proteínas: el polimorfismo Kozak incrementa en las plaquetas la densidad del receptor glucoproteico Ib alfa27 y el VNTR produce la prolongación del dominio de unión del ligando-receptor de FvW, la prolongación del dominio dentro del torrente sanguíneo es mayor, lo que probablemente altera la activación de las plaquetas por distintos inductores y la susceptibilidad de éstas para unirse con el FvW.28
El polimorfismo del receptor de ADP
Recientemente, Fontana describió el polimorfismo del receptor P2Y12 que modula la reactividad plaquetaria.29 El haplotipo H2 del gen que codifica este receptor se correlaciona con el efecto agregante máximo de las plaquetas en respuesta al ADP y se demostró que se asocia con enfermedad arterial periférica.29,30
Métodos de laboratorio para identificar el síndrome de hiperactividad de las plaquetas
Una amplia variedad de pruebas de laboratorio de la función plaquetaria se encuentran disponibles en la actualidad,31 pero no todas han sido utilizadas para estudiar la hiperactividad plaquetaria (Tabla 1).
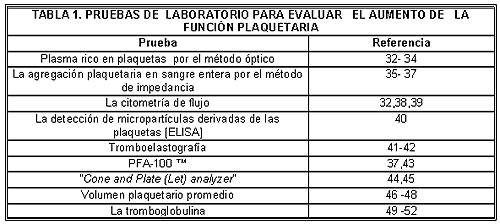
El aumento de la agregación plaquetaria ha sido definido clásicamente utilizando el método óptico introducido por Born.53 Este es un estudio engorroso que requiere la centrifugación como primer paso, que puede producir la activación y posiblemente el agotamiento de las plaquetas. Además, excluye de la evaluación las plaquetas más grandes, más pesadas y –posiblemente– las hiperactivas.54 A su vez, no incluye adecuadamente los glóbulos rojos y blancos que modulan significativamente la función de las plaquetas.55 Recientemente, la introducción de los estudios de impedancia de la agregación plaquetaria en sangre entera ha aportado ventajas en relación con el método óptico, entre las que figura la evaluación rápida de la función de las plaquetas sin centrifugación y en condiciones similares a las fisiológicas.56 Debido a que la respuesta funcional de las plaquetas a algunos agonistas y otras drogas depende de la presencia de eritrocitos y leucocitos,57 la sangre entera es superior al PRP, en condiciones fisiológicas, para poder evaluar el efecto de las drogas sobre la función y la reactividad plaquetarias. La sensibilidad elevada de las pruebas en sangre entera permite medir la agregación espontánea y emplear distintas concentraciones de agonistas (ácido araquidónico, colágeno y ADP) que evalúan la reactividad de estas células.
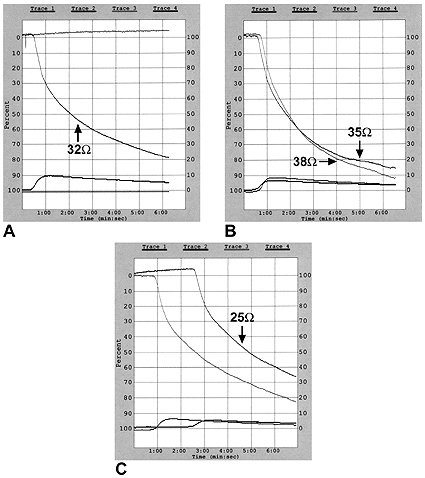
Figura 1. Niña de 15 años a la que se le diagnosticó trombosis de la vena mesentérica superior. Los trazados A, B, C muestran el aumento de la respuesta agregante a diferentes concentraciones de ácido araquidónico (AA) en la sangre entera; A: 0.5 mM de AA, B: 0.25 mM y 0.125 mM de AA, C: 0.063 mM de AA.
La Figura 1 muestra los trazados de la agregación plaquetaria en una niña de 15 años con historia de trombosis en la vena mesentérica superior. Los estudios de agregación plaquetaria comprobaron SHP. Se observó que el ácido araquidónico indujo el aumento de la actividad plaquetaria y que las concentraciones más bajas de este ácido (0.25; 0.125 mM) desencadenaron en éstas una respuesta agregante más fuerte (38 y 35 ohms, respectivamente) que las concentraciones más altas del agonista (0.5 mM; 32 ohms).
La hiperactividad de las plaquetas se caracteriza por una respuesta agregante elevada ante concentraciones bajas del agonista y por la agregación espontánea. Sin embargo, el diagnóstico debe realizarse sobre la base de ambos, los hallazgos de laboratorio que demuestran el aumento en la agregación plaquetaria y la historia clínica del paciente.
Además de las pruebas de agregación plaquetaria se investigó la utilidad de la citometría de flujo para evaluar el aumento de la agregación.58 Este estudio permite medir la expresión de la selectina P,13,59 la unión del fibrinógeno,60,61 y la liberación de micropartículas que derivan de las plaquetas,62 así como los agregados de plaquetas y monocitos.63,64 Desafortunadamente, es considerada solamente como una herramienta de investigación, debido a que es más cara que la impedancia de la agregación plaquetaria en sangre entera y a que la preparación de las muestras requiere más tiempo.65 Desde el punto de vista práctico, la prueba de la agregación plaquetaria en sangre entera aparenta ser el método de primera elección porque es más rápido y evalúa con precisión el aumento de la función en estas células.
Los desafíos en el tratamiento del síndrome de hiperactividad de las plaquetas
Los pacientes con SHP que presentan complicaciones tromboembólicas, frecuentemente recurrentes, requieren un tratamiento efectivo que inhiba la función de las plaquetas. Se demostró que la aspirina inhibe la función de éstas en los pacientes con este síndrome.5 No obstante, recientemente también se describió la resistencia a la aspirina en la enfermedad cardiovascular y cerebrovascular.66-68 Un segundo enfoque incluye el uso de agentes antiagregantes plaquetarios más recientes como el clopidogrel; sin embargo, existen datos que reflejan la falta de respuesta a este derivado de la tienopiridina.69,70 Es por esto que la hiperactividad de las plaquetas representa uno de los desafíos más importantes para lograr un tratamiento efectivo que inhiba la función de las plaquetas.69
Los desafíos principales se observan en los pacientes con el polimorfismo PlA2 en la glucoproteína IIIa. Estos no presentan hiperactividad de las plaquetas exclusivamente13-15 y algunos estudios demostraron que estos pacientes pueden presentar resistencia a la aspirina.73 Los portadores del alelo PlA2 con enfermedad cardíaca coronaria pueden beneficiarse con el tratamiento con clopidogrel;72 sin embargo, la observación de pacientes sometidos a intervenciones coronarias no ha confirmado esta hipótesis.73 A su vez, el polimorfismo P2Y12 del receptor puede afectar la respuesta al clopidogrel. En los pacientes con el haplotipo H2 en el gen que codifica este receptor, la respuesta agregante al ADP puede estar aumentada y estos individuos pueden presentar resistencia al clopidogrel.29 Por este motivo, la farmacogenomia moderna es una herramienta potencial para decidir el tratamiento óptimo para el paciente individual y superar la resistencia a las drogas.74
Hasta el momento, no se han establecido guías de tratamiento para el SHP. No obstante, sobre la base de nuestras observaciones y experiencia, la aspirina en dosis de 325 mg podría ser la terapia de primera línea en el SHP de diagnóstico reciente. Si estas dosis son ineficaces, el médico puede duplicarlas. Si con esta droga no se inhibe el funcionamiento de las plaquetas, el paciente puede recibir una terapia dual con aspirina y clopidogrel. El clopidogrel debe usarse en forma aislada en los casos en que la aspirina esté contraindicada (hipersensibilidad a la aspirina, asma inducida por aspirina). En todos los pacientes con SHP que se encuentran en tratamiento con antiagregantes se debe controlar la función de las plaquetas y la respuesta a las drogas.
Trombocitosis y trombocitemia
Todas las formas de los síndromes mieloproliferativos (leucemia mieloide crónica, policitemia vera, mielofibrosis idiopática y trombocitemia esencial) tienen el potencial para producir trombocitemias de tipo clonal. En cada uno de estos síndromes, la expresión progresiva de la masa de megacariocitos y el incremento resultante en el número de plaquetas provee la base para la aparición de los síntomas. Como entidad clínica, la trombocitemia esencial representa el cuadro clásico que se caracteriza por sucesos clínicos y terapéuticos relacionados con las alteraciones en el número y función de las plaquetas. Es común que se correlacione con la hiperactividad plaquetaria y, menos frecuentemente, con plaquetas hipoactivas, lo que hace que el foco se centre en el número aumentado de plaquetas que desempeñan un papel importante en los trastornos clínicos como la trombosis y las hemorragias.
Trombocitosis
La trombocitosis es un hallazgo de laboratorio frecuente en la práctica clínica. Es común que sea descubierto accidentalmente cuando se solicita el recuento sanguíneo completo por una causa no relacionada. El desafío diagnóstico para el médico lo constituye el hecho de reconocer aquellos pacientes con trombocitemia que habitualmente sufren episodios trombohemorrágicos, de aquellos individuos con seudotrombocitosis y trombocitosis reactivas y que no presentan riesgo elevado para dichos episodios.
El término “seudotrombocitosis” se aplica a los sujetos con cualquier forma de crioglobulinemia en la que se identifica un recuento elevado de plaquetas secundario a artefactos del conteo electrónico. Las proteínas circulantes precipitan y se interpretan erróneamente como partículas del mismo tamaño que las plaquetas. El examen de frotis sanguíneo corrige este error. Como es esperable, el reconocimiento de los sucesos trombohemorrágicos no se correlaciona con el reconocimiento o con la enumeración de tales precipitados.75
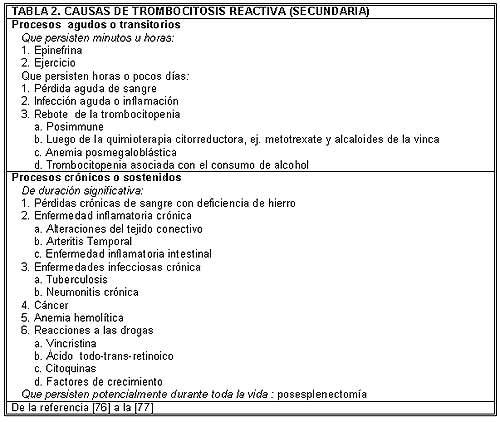
La “trombocitosis reactiva o secundaria” define a los pacientes con un recuento elevado del número de plaquetas que presentan alguna condición clínica asociada frecuentemente con dicho aumento. La Tabla 2 enumera las causas más comunes de trombocitosis reactiva. En general, la trombocitosis reactiva no se asocia con el incremento de la incidencia en el riesgo de las manifestaciones trombohemorrágicas, excepto en los casos en los que existe una enfermedad hematopoyética subyacente con características protrombóticas, lesiones arteriales preexistentes significativas o inmovilidad prolongada.75,78
El término “trombocitemia” se aplica a los estados mieloproliferativos en los que existe estimulación autónoma de la producción de plaquetas que lleva al aumento en el número de estas células. Estas lesiones son secundarias a la expansión clonal de una célula pluripotencial hematopoyética con tendencia marcada a la transición de una forma a otra durante el curso clínico.79 Los mecanismos exactos para la expresión específica de este linaje, la amplificación y la proliferación de cada forma, excepto los casos de leucemia mieloide crónica, no quedan claros hasta el momento.
Ruggeri y col.80 realizaron un estudio transversal y prospectivo en una cohorte de 10 000 personas sanas para estimar la frecuencia de policitemia vera (PV) o trombocitemia esencial (TE) en personas con eritrocitosis o trombocitosis, en la población general. Al comienzo, se observó una prevalencia mayor de la esperada para PV y TE. Luego de 5 años de seguimiento, sólo una persona de los 99 casos con trombocitosis al inicio del estudio desarrolló TE y no se registraron casos adicionales de PV. Es por esto que el riesgo de contraer trombocitemia en personas con trombocitosis es bajo.
Los mecanismos de la trombocitosis
La trombopoyetina (TPO) regula la diferenciación y proliferación megacariocítica y la interleuquina 6 (IL-6) y la IL-11 podrían desempeñar un papel accesorio en este proceso.81 La TPO plasmática se une al receptor c-Mpl en la superficie de las plaquetas circulantes; la TPO libre queda disponible para promover la proliferación megacariocítica en la médula ósea. Por esto, en los estados trombocitopénicos, la disminución del recuento de plaquetas conduce al aumento de los niveles de TPO libre en plasma que estimula la megacariocitopoyesis. En la trombocitosis, el aumento del recuento de plaquetas provoca el descenso de los niveles de TPO libre en plasma con reducción de la megacariocitopoyesis.
En la trombocitosis reactiva secundaria a un proceso inflamatorio subyacente los niveles de TPO pueden ser altos y pueden anteceder al incremento del número de plaquetas.82,85 La IL-6 podría mediar el aumento de la producción de la TPO en el hígado ya que regula positivamente el incremento del ARN mensajero de esta hormona en dicho órgano.86,87
En la trombocitemia o trombocitosis clonal, los niveles de TPO pueden estar aumentados.88 Sin embargo, esto puede ser debido a un defecto en las plaquetas o a la expresión del c-Mpl en los megacariocitos y las plaquetas, que lleva al deterioro de la unión de la TPO y que los niveles libres en plasma sean mayores a los esperados. El incremento de los niveles de la TPO en pacientes con TE también puede deberse al aumento del receptor soluble de IL-6, un agonista de la actividad de esta citoquina.89 Junto con el aumento de la sensibilidad a la TPO en los megacariocitos de los pacientes con TE, los altos niveles de TPO libre conducen al aumento notable del recuento de plaquetas que se observa en estos individuos.90-94 La disminución de la sensibilidad a la actividad supresora del factor transformador del crecimiento β1 (FCT-β1) en las unidades formadoras de colonias megacariocíticas, de los individuos con TE, también puede contribuir al descontrol de la megacariocitopoyesis.95
Trombocitemia esencial
Los sucesos trombohemorrágicos son las principales manifestaciones clínicas de la trombocitemia esencial (primaria). En Estados Unidos este término se aplica a las alteraciones clonales de los precursores de los megacariocitos, y –recientemente- en Europa se la ha denominado “trombocitemia vera”96 que ayuda a definir esta patología como una condición que va más allá del incremento en el número de plaquetas.
Los criterios diagnósticos de la trombocitemia esencial
Los criterios diagnósticos actuales de la clasificación de tumores de la OMS97 se enumeran en la Tabla 3. Los criterios originales fueron delineados por The Polycythemia Vera Study Group (PVSG), en 1975,98 y luego se modificaron a medida que mejoró la comprensión del proceso de la enfermedad.99-105 La formación espontánea de colonias megacariocíticas es un criterio adoptado con entusiasmo en Europa,96 aunque en Estados Unidos es infrecuente que se realice su determinación, debido a que los requerimientos de laboratorio son complejos. Se debe efectuar el examen morfológico detallado de la sangre periférica y del material de médula ósea para excluir otros trastornos mieloproliferativos y estados mielodisplásicos que cursan con trombocitosis.

En los últimos 4 años se publicaron distintos estudios acerca del uso de la cuantificación del ARN mensajero del gen de la policitemia rubra vera del tipo 1 (PRV-1) de los granulocitos como marcador diagnóstico de las enfermedades mieloproliferativas, entre las que se incluyen la PV y la TE.106-110 El PRV-1 es un receptor de superficie de células hematopoyéticas, un nuevo miembro de la superfamilia del receptor activador de plasminógeno del tipo uroquinasa (uPAR).111 Aunque existen aspectos metodológicos por resolver,109 el aumento de la expresión de PRV-1 en los granulocitos podría convertirse en una herramienta diagnóstica complementaria para poder distinguir entre pacientes con policitemia de aquellos con eritrocitosis y entre individuos con trombocitemia de aquellos con trombocitosis secundaria.113,114 Este aumento en la expresión también podría identificar un subgrupo de individuos con TE y tendencia a presentar PV o sucesos trombohemorrágicos.106
Características clínicas
La verdadera incidencia de la TE se desconoce, pero la incidencia estimada varía entre 1-2.5 casos por 100 000 individuos por año.115 La mayoría de los casos se producen entre la sexta y séptima décadas de la vida, con la misma distribución por sexo. Un segundo pico de incidencia tiene lugar cerca de los 30 años y es más frecuente que afecte a las mujeres que a los hombres.115-117 La TE puede presentarse en niños de hasta 9 meses, aunque es raro.118
Las lesiones trombóticas son las características clínicas predominantes de la TE. Estas comprenden desde episodios isquémicos transitorios en la retina, sistema nervioso central y corazón hasta el espectro completo de síntomas secundarios a la disminución del flujo sanguíneo por oclusión vascular que puede causar angina, infarto de miocardio, accidente cerebrovascular y trombosis de la vena porta. La heterogeneidad de los sitios afectados y la ausencia de otros mecanismos característicos de trombosis deben sugerir el diagnóstico de TE. Dos aspectos clínicos específicos de TE deben ser descritos con mayor detalle, debido a que facilitan la comprensión de los eventos fisiopatológicos: la eritromelalgia y la trombosis de la microcirculación.
La eritromelalgia aparenta ser un hallazgo patognomónico en pacientes con trobocitemia primaria, aunque ésta también puede estar presente en los casos de PV con trombocitemia asociada.119,120 La eritromelalgia presenta un perfil clínico característico que permite que se diagnostique rápidamente mediante el interrogatorio. Es frecuente que comience con acroparestesias o prurito en los dedos de los pies. A éstos les siguen el dolor y la quemazón en las yemas de los dedos de los pies y es común que estos últimos y el pie estén rojos y congestivos. El dolor puede ser muy intenso y el calor o el ejercicio pueden desencadenarlo.119 Estos episodios ocurren incluso con la elevación marginal del número de plaquetas. Se han identificado cambios histopatológicos significativos que se caracterizan por la presencia de cambios vasculares en las arteriolas, con edema de las células endoteliales y proliferación fibromuscular interna.121 A través de las investigaciones exhaustivas de Michiels y col. se demostró que la eritromelalgia se produce por la inflamación acral y la trombosis arteriolar mediada por las plaquetas. El aspecto más importante de esta lesión es la respuesta inmediata del complejo de síntomas a la administración de aspirina.122,123
Se debe poner énfasis en la terminología que se aplica a esta secuencia fisiopatológica. Los estudios de Michiels y col.96,120-122 proveen el fundamento para restringir el término eritromelalgia a las circunstancias de la trombocitemia en las que la biología y la terapia aparentan ser específicas. En contraste, el término eritromalgia es más apropiado para ser aplicado al conjunto de trastornos circulatorios, inflamatorios o neurológicos en los que se presentan cambios vasculares secundarios (lupus, artritis reumatoidea, endarteritis obliterante, diabetes).
La trombosis de la microcirculación abarca las lesiones oclusivas arteriales y arteriolares que producen una amplia gama de síntomas entre los que figuran los episodios isquémicos transitorios, las alteraciones de la visión que se producen por la oclusión de los vasos de la retina, la claudicación intermitente y los infartos digitales.118,124 Es probable que éstas sean las manifestaciones más frecuentes de la trombocitemia. Es común que las oclusiones vasculares sean asintomáticas debido a que se ubican en sitios que no se manifiestan y esto dificulta su reconocimiento.
Históricamente, se consideró infrecuente la oclusión de los grandes vasos en la trombocitemia primaria. Debido a que la comprensión de esta entidad clínica ha mejorado, queda claro que la incidencia de la enfermedad oclusiva arterial o coronaria es alta. Además, es notable la incidencia significativa de la oclusión venosa mesentérica, quizá la causa más frecuente del síndrome de Budd-Chiari. Griesshammer y col.122 revisaron 11 estudios clínicos retrospectivos con 809 pacientes y demostraron que resultaba sorprendente el gran número de casos con trombosis arterial cerebral, de la vena porta (asociada al síndrome de Budd-Chiari) y trombosis arterial coronaria. Por este motivo, a pesar de que en el pasado se hacía hincapié en las alteraciones de la microcirculación, queda claro que se pueden comprometer vasos de mayor calibre. El priapismo es una forma especial de presentación de las trombosis del lecho vascular intermedio.
En los pacientes con TE la hemorragia tiene lugar con menor frecuencia que la trombosis y es raro que sea grave. Lo más común es que ésta ocurra en las superficies mucosas. Son habituales los antecedentes de epistaxis, gingivorragia y hemorragia gastrointestinal leve o genitourinaria. También son comunes los hematomas con superficie equimótica, especialmente en las extremidades. Pueden tener lugar cambios visuales importantes debido a hemorragias retinianas espontáneas, las cuales representan una complicación potencialmente grave.129 La hemorragia aparenta ser más frecuente y significativa en la trombocitemia que se asocia con otras lesiones mieloproliferativas que en la trombocitemia primaria y también en pacientes cuyo recuento de plaquetas es mayor que 1.5 millones/μl.101,130-133
En mujeres con trombocitemia se observa una alta incidencia de complicaciones obstétricas. Es común la trombosis de los vasos de la placenta, con infartos placentarios y abortos relacionados. Cabe destacar que la declinación espontánea en el número de plaquetas es habitual durante el curso del embarazo. También se puede esperar que el embarazo llegue a término en aquellas pacientes en las que la agregación plaquetaria se controla con aspirina o en las que el número de plaquetas se reduce con la administración de interferón.134-137
La esplenomegalia de grado leve está presente en 50% a 70% de los pacientes en el momento del diagnóstico, y la hepatomegalia, en 15% a 20%.125-128 En 30% de los pacientes con TE se observa hipertensión y puede ser una de las enfermedades asociadas que aumenta el riesgo de lesiones vasculares oclusivas.
Fisiopatología de la trombocitemia esencial
En las personas con trombocitemia primaria tiene lugar una variedad de situaciones fisiopatológicas interesantes. En primer término, los cambios endovasculares en los individuos con eritromelalgia han sido bien identificados. Estos cambios abarcan el estrechamiento luminal nuclear y extenso debido a la proliferación de las células del músculo liso con formación de vacuolas y edema en su citoplasma, depósitos de material intercelular y fragmentación de la lámina elástica interna.120,121 En segundo lugar, se detectaron cambios en la arquitectura y función de las plaquetas que podrían estar relacionados con los síntomas clínicos y los cambios endoteliales descritos. Estos cambios incluyen el tamaño heterogéneo de las plaquetas y variaciones ultraestructurales, niveles elevados de proteínas plaquetarias específicas, el aumento de la producción de tromboxano y la expresión de epitopes dependientes de la activación en la superficie de las plaquetas.133 El tercer hallazgo se relaciona con la elevación de los niveles de trombopoyetina88 descrita con anterioridad. En una de las formas familiares de trombocitemia primaria se encontraron niveles elevados de tromboyetina en los miembros afectados y la deleción en uno de los pares de bases en la región 5’ no transcripta del gen de la TPO.133 Por lo menos en una de las formas familiares el cambio genético parece ser importante en la regulación de la expresión de la TPO y representa la base de la forma familiar de la trombocitemia. En Japón, un informe reciente demostró que en una estirpe con trombocitemia esencial familiar, la mutación activadora dominante- positiva del gen cMpl es la responsable de la enfermedad138 y simula la actividad aumentada de la TPO. Por último, en pacientes con trombocitemia se vio una relación inversa entre los recuentos plaquetarios elevados y los multímeros grandes del factor de Von Willebrand (FvW). El incremento del número de plaquetas se relaciona con el aumento de la degradación de los multímeros del FvW unidas a las plaquetas.130 Se considera que estos efectos son un factor adicional para las alteraciones de la hemostasia en la trombocitemia.
Fisiopatología de la trombosis en la trombocitemia esencial
Mientras que la trombosis es la manifestación clínica predominante en la TE, es poco frecuente en la trombocitosis reactiva, lo que sugiere que el aumento aislado en el número de plaquetas no es suficiente para explicar la alta incidencia de eventos trombóticos. Aunque el recuento descontrolado de plaquetas es un factor de riesgo para la trombosis,39-142 Regev y col.143 documentaron que el 70% de sus pacientes presentaron síntomas vasculares y recuentos plaquetarios menores de 600 000/μl y que en el 50% eran menores de 500 000/μl.
En la TE se observan alteraciones en la arquitectura de las plaquetas y los megacariocitos, con plaquetas de forma anormal y gran tamaño en la circulación. Sin embargo, las investigaciones no han podido relacionar estos cambios estructurales con el riesgo aumentado de trombosis.75,144-147 De manera similar, los trabajos de función plaquetaria no pudieron identificar las alteraciones correlativas que podrían ser utilizadas para definir el riesgo de trombosis. Sin embargo, la aplicación reciente del método de agregación plaquetaria en sangre entera pudo reconocer la hiperactividad significativa de estas células in vitro en pacientes con TE y antecedentes de trombosis.148 El uso de sangre entera contrasta con el método óptico de agregación plaquetaria convencional, que define mejor la hipoactividad de las plaquetas. Estudios previos acerca de la interacción de las plaquetas con el endotelio,149 el incremento del factor 4 plaquetario (FP4) y los niveles de tromboglobulina β (TGβ) aportan datos que demuestran que la activación excesiva de estas células en la TE es un mecanismo potencial del aumento de la trombosis.149-152 No obstante, el riesgo de trombosis se correlaciona mejor con la edad que con el FP4 y la TGβ.103,153 Otros intentos para examinar los parámetros que se relacionan con el endotelio, como los niveles de homocisteína, no han logrado comprobar esta relación,154 aunque la derivación reciente de una línea celular de un individuo con TE mostró la combinación de características endoteliales y hematopoyéticas que indica una relación ontogenética estrecha entre estos linajes.155
La hiperactividad plaquetaria podría ser explicada en parte a través del incremento en el factor de crecimiento del endotelio vascular (FCEV)156 y la trombopoyetina,88 que ha sido descrito en las personas con TE y PV. Se demostró la activación de las plaquetas durante la estimulación de estos factores.158 Cacciola y col.159 estudiaron la relación del FCEV con la TPO en 45 sujetos con TE y 25 con PV, que no habían experimentado acontecimiento trombótico alguno antes de la investigación. Aquellos que presentaron trombosis tenían valores basales más altos del FCEV. Así, los niveles altos de este factor podrían servir para reconocer los pacientes con riesgo aumentado de trombosis.
Mientras que en los sujetos con TE se observan estados trombofílicos genéticos y adquiridos (factor V de Leiden, protrombina G20210A, metilentetrahidrofolato reductasa C677T, homocisteinemia, deficiencia de proteínas C y S, deficiencia de antitrombina y anticuerpos anticoagulante lúdico y antifosfolipídico), en estos casos no existe correlación entre estos factores protrombóticos con la aparición de trombosis. Por lo que la detección sistemática rutinaria de estas condiciones no se justifica en la TE.160
La historia natural de la trombocitemia esencial
La caracterización y descripción del curso natural de la TE han sido obstaculizados en virtud de la falta de acuerdo absoluto en los criterios diagnósticos, el sesgo en la selección de la población (datos provenientes de unidades específicas de trombosis), diferencias importantes respecto de los parámetros e indicaciones para el tratamiento y períodos de seguimiento limitados de una enfermedad que dura décadas.
En estudios con pequeñas cohortes de enfermos no han surgido datos claros que muestren diferencias sustanciales en la expectativa de vida entre los que presentan TE y controles apareados por edad y sexo.161-164 Esto ha llevado a que la TE no sea considerada una enfermedad. No obstante, cuando se observa una serie de casos con una presentación bien definida, esto demuestra que puede ser necesaria una nueva interpretación de este enfoque. Uno de los mejores trabajos, realizado en Turín, Italia, por Bazan y col., llevó a cabo el seguimiento consecutivo de 187 pacientes. El 50% de éstos presentó un episodio trombótico dentro de los 9 años del diagnóstico. No se observaron diferencias significativas en las curvas de supervivencia libre de trombosis o en la supervivencia global en menores o mayores de 50 años de edad y sólo el 85% estaba vivo a los 10 años del diagnóstico. Estos autores demostraron que la tasa relativa de mortalidad era 4 veces mayor en las personas con TE al compararlas con los controles sanos apareados por edad y por sexo. Lo interesante fue que otros factores comunes combinados, expresados como variables de confusión en la evaluación del efecto de TE sobre el riesgo cardiovascular (edad en el momento del diagnóstico, hábito de fumar, sexo, hipercolesterolemia, hipertensión y diabetes) no fueron significativos, como tampoco el número máximo de plaquetas.
Debido a que la TE es un tipo de síndrome mieloproliferativo, no es sorprendente que la progresión hacia la PV o la mielofibrosis idiopática crónica (MIC) se observe en un porcentaje de pacientes cercano al 5%,132,162,163 aunque estas cifras derivan de trabajos pequeños. La progresión hacia la leucemia mieloide aguda se registró en 3% a 4% de los sujetos con TE,132 pero todos habían estado expuestos a agentes leucemogénicos potenciales que se utilizaron para controlar el número de plaquetas. En sujetos tratados con agentes mielosupresores (busulfán, hidroxiurea) también se describió el incremento en la incidencia del síndrome mielodisplásico y de tumores sólidos.165
El tratamiento de la trombocitopenia esencial
El reconocimiento de la TE en personas sin otras enfermedades y los indicios que sugieren que algunas maniobras simples, como la administración de aspirina, pueden mejorar la expectativa de vida libre de trombosis135,166 sustenta el concepto de que la planificación del tratamiento debe realizarse sobre la base de la estratificación de riesgo.135,139,140,142,166-169 Esta estratificación se relaciona ampliamente con los factores que modifican las complicaciones trombohemorrágicas. En la Tabla 4, Tefferi169 enumera parámetros simples para dicha estratificación en la planificación del tratamiento. Por cierto, se debe revisar si el aumento de la expresión del PRV-1 y de los niveles del FCEV se deben incorporar en dicha estratificación y pueden ser necesarios estudios prospectivos para examinar si éstos son realmente factores de riesgo para eventos trombohemorrágicos.106,170 El enfoque para tratar a los pacientes con TE se ha centrado en la administración de agentes inhibidores de la agregación plaquetaria y en la reducción en el número de plaquetas circulantes.

Tradicionalmente, el interés por la utilización de agentes inhibidores de la agregación plaquetaria se ha centrado en las investigaciones que mostraron que la disfunción plaquetaria es frecuente en la trombocitemia primaria. A su vez, parte del fenómeno hemorrágico ha sido atribuido a este defecto sobre la función de las plaquetas. Estos hallazgos hacen que se dude de cualquier agente que pueda afectar dicha función. Los datos que indicaron que la aspirina puede revertir las lesiones vasculares, incluso en casos de gangrena incipiente,124 hicieron que esta tesis sea cuestionada. Recientemente se constató que el tratamiento con aspirina mejora la función neurológica, incluso cuando persiste el aumento en el número de plaquetas.129 Resulta claro que este fármaco es efectivo en la reducción de los síntomas y signos de las personas con lesiones vasculares oclusivas.122,123,125,171 Además, esta droga se ha convertido en un agente terapéutico importante durante el embarazo.125,135-137,146 Quedó demostrado que la aspirina es efectiva para ayudar a que el embarazo llegue a término satisfactoriamente. En la actualidad, la experiencia con nuevos agentes inhibidores de la agregación plaquetaria como el clopidogrel es escasa para poder definir el papel que desempeñan en estas mujeres.
Además de los agentes inhibidores de la agregación plaquetaria se utilizan varias estrategias para reducir el número de plaquetas. La plaquetaféresis disminuye rápidamente el recuento de plaquetas (en horas). Desafortunadamente, su efecto es de corta duración (de horas a días). No se la utiliza frecuentemente pero su valor se asienta en que puede revertir rápidamente las lesiones trombóticas de la microvasculatura o en circunstancias en las que se requiere cirugía de urgencia.
Se han utilizado casi todas las drogas mielosupresoras citorreductoras (fósforo, agentes alquilantes, antimetabolitos). Estos agentes pueden brindar resultados satisfactorios y reducir el número de plaquetas. Lamentablemente, la mayoría son potencialmente leucemogénicos, de manera tal que se los ha abandonado en favor de la hidroxiurea.
La hidroxiurea se ha convertido en el agente citorreductor de primera elección. Esta mólecula simple se sintetizó hace más de 100 años, pero recién en 1960 se introdujo en la medicina clínica cuando se demostró que inhibía la ribonucléotido reductasa.172-174 La ausencia de toxicidad con este agente es relativa y presenta un amplio margen dosis-respuesta. Induce cambios megaloblásticos esperables relacionados con su sitio de acción.175 El entusiasmo para su uso se fundamentó en la creencia de que no era leucemogénico; sin embargo, se podría considerar un agente con potencial cancerígeno incierto ya que se ha vinculado este tratamiento con la evolución de la trombocitemia primaria a leucemia aguda.172,176,177
En la mayoría de los sujetos con trombocitopenia primaria, con una dosis diaria de 500 a 1 000 mg/día por vía oral, se podrá constatar un descenso del número de plaquetas, para luego ajustar las dosis, de manera tal que se pueda obtener el número de plaquetas circulantes esperado.75,140,178,179
El interferón demostró que puede disminuir con efectividad el recuento plaquetario en pacientes con trombocitemia primaria o trombicitemia asociada con otros trastornos mieloproliferativos.101,180-185
Su mecanismo de acción podría estar mediado por el efecto inhibidor de la megacariopoyesis. En la actualidad, su uso se reserva a las embarazadas debido a sus efectos colaterales, el esfuerzo que demanda la administración por vía parenteral y su costo. Su valor reside en que se puede evitar el uso de agentes potencialmente mutagénicos. La dosis terapéutica habitual es de 3 millones de UI por vía subcutánea 3 veces por semana. Con el tratamiento con interferón, cerca del 80% al 90% de los pacientes tratados obtienen la remisión. Al interrumpir el tratamiento, es usual que recurran los sucesos clínicos y de laboratorio. La remisión se mantiene en alrededor del 10% de los pacientes.
Luego de casi una década de ensayos clínicos, la anagrelida se suma a las herramientas terapeúticas para los individuos con trombocitemia, sin considerar la forma subyacente de enfermedad mieloproliferativa. Su introducción se produjo debido a que posee actividad inhibidor de la fosfodiesterasa del monofosfato cíclico de adenosina e inhibe la agregación plaquetaria.186 Los datos que avalan la reducción en el número de plaquetas dependiente de la dosis en controles sanos187 y en pacientes con trombocitemia, hacen que su uso sea considerado una modalidad terapéutica importante. Su mecanismo de acción no está completamente identificado, pero en trabajos in vivo e in vitro se vio que altera la maduración de los megacariocitos, con disminución de las alteraciones en su tamaño y morfología.192,193 Cabe destacar que otros hallazgos, como la supervivencia de las plaquetas y la proliferación de la reserva progenitora de los megacariocitos fueron normales. En general, la dosis inicial puede ser de de 0.5 a 1 mg administrada 2 o 4 veces por día. El descenso significativo en el número de plaquetas es evidente desde los 7 hasta los 14 días, momento en el cual se puede ajustar la posología. Las dosis individuales no deben superar los 2 mg y se recomienda que la dosis máxima sea de 10 mg/día.189 Es raro que se requieran más de 4 mg por día. En el presente se realizan ensayos clínicos con una forma de anagrelida con efecto prolongado y que podría simplificar el esquema de tratamiento.
La anagrelida presenta efecto vasodilatador y se observaron efectos colaterales que incluyen cefalea (cerca del 50%), hipotensión postural, retención de fluidos y diarrea. En un porcentaje significativo de pacientes (cercano al 70%)194 Las palpitaciones y la taquicardia observadas aparentan ser peores en los que reciben dosis cercanas a la máxima. Estos síntomas desaparecen luego de 4 a 8 semanas del tratamiento. Se demostró que el uso prolongado de la anagrelida en individuos jóvenes es seguro y efectivo y que la anemia moderada o leve fue el único efecto colateral registrado luego de un tiempo promedio de seguimiento de 10.8 años.195
A pesar de que es costosa, esta droga aportó un avance terapéutico notable en el enfoque de la trombocitemia.
Conclusiones
La hiperactividad de las plaquetas desempeña un papel importante en la patogenia de la trombosis y puede ser causa de episodios tromboembólicos recurrentes. La comprensión apropiada del significado clínico del aumento de la función de las plaquetas ha llevado a que se establezcan nuevas pruebas funcionales en condiciones similares a las fisiológicas, que son útiles para evaluar las “plaquetas hiperactivas”. El uso del método de impedancia de la agregación plaquetaria en sangre entera permitió que se reformulen las características de laboratorio del síndrome de hiperactividad de las plaquetas y aportó datos relacionados con el aumento de la función de estas células en los síndromes mieloproliferativos. El enfoque actual de las alteraciones plaquetarias en la trombocitemia se centra en la terapia combinada de drogas antiplaquetarias y agentes que controlan el número de estas células para evitar la aparición de las complicaciones trombóticas que caracterizan esta entidad clínica.
Los autores no manifiestan “conflictos de interés”.
Bibliografía del artículo
- Heemskerk JW, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost. 2002; 88:186-93.
- Rao AK. Congenital disorders of platelet function: disorders of signal transduction and secretion. Am J Med Sci. 1998; 316:69-76.
- White JG, Key NS, King RA et al. The White platelet syndrome: a new autosomal dominant platelet disorder. Platelets. 2004; 15:173-84.
- Fujimoto TT, Sora M, Ide K et al. Glanzmann thrombasthenia associated with a 21-amino acid deletion (Leu817-Gln837) in glycoprotein IIb due to abnormal splicing in exon 25. Int J Hematol. 2004; 80:83-90.
- Mammen EF, Barnhart MI, Selik NR et al. “Sticky platelet syndrome”: a congenital platelet abnormality predisposing to thrombosis Folia Haematol Int Mag Klin Morphol Blutforsch 1988; 115:361-5.
- Mammen EF. Sticky platelet syndrome. Semin Thromb Hemost. 1999; 25:361-5.
- Rubenfire M, Blevins RD, Barnhart M et al. Platelet hyperaggregability in patients with chest pain and angiographically normal coronary arteries. Am J Cardiol. 1986; 57:657-60.
- Holiday PL, Mammen EF, Gilroy J et al. Sticky platelet syndrome and cerebral infarction in young adults. Ninth Int Joint Conf on Stroke and Cerebral Circulation. Phoenix, AZ; 1983.
- Mammen EF. Ten years’ experience with the “sticky platelet syndrome”. Clin Appl Thromb Hemost 1995;1:66-72.
- Bick RL. Sticky platelet syndrome: a common cause of unexplained arterial and venous thrombosis. Clin Appl Thromb Hemost 1998; 4:77-81.
- Andersen JA. Report: bleeding and thrombosis in women. Biomed Progress 1999; 12:40.
- Weber M, Gerdsen F, Gutensohn K, Schoder V et al. Enhanced platelet aggregation with TRAP-6 and collagen in platelet aggregometry in patients with venous thromboembolism. Thromb Res. 2002; 107:325-8.
- Michelson AD, Furman MI, Goldschmidt-Clermont P et al. Platelet GP IIIa Pl(A) polymorphisms display different sensitivities to agonists. Circulation. 2000; 101:1013-8.
- Feng D, Lindpaintner K, Larson MG et al. Increased platelet aggregability associated with platelet GPIIIa PlA2 polymorphism: the Framingham Offspring Study. Arterioscler Thromb Vasc Biol. 1999; 19:1142-7.
- Goodall AH, Curzen N, Panesar M et al. Increased binding of fibrinogen to glycoprotein IIIa-proline33 (HPA-1b, PlA2, Zwb) positive platelets in patients with cardiovascular disease. Eur Heart J. 1999; 20:742-7.
- Gardemann A, Humme J, Stricker J et al. Association of the platelet glycoprotein IIIa PlA1/A2 gene polymorphism to coronary artery disease but not to nonfatal myocardial infarction in low risk patients. Thromb Haemost. 1998; 80:214-7.
- Mikkelsson J, Perola M, Laippala P et al. Glycoprotein IIIa Pl(A) polymorphism associates with progression of coronary artery disease and with myocardial infarction in an autopsy series of middle-aged men who died suddenly. Arterioscler Thromb Vasc Biol. 1999; 19:2573-8.
- Ridker PM, Hennekens CH, Schmitz C et al. PIA1/A2 polymorphism of platelet glycoprotein IIIa and risks of myocardial infarction, stroke, and venous thrombosis. Lancet. 1997; 349:385-8.
- Kunicki TJ, Kritzik M, Annis DS et al. Hereditary variation in platelet integrin alpha 2 beta 1 density is associated with two silent polymorphisms in the alpha 2 gene coding sequence. Blood.1997; 89:1939-43.
- Santoso S, Kunicki TJ, Kroll H et al. Association of the platelet glycoprotein Ia C807T gene polymorphism with nonfatal myocardial infarction in younger patients. Blood. 1999; 93:2449-53.
- Carlsson LE, Santoso S, Spitzer C et al. The alpha2 gene coding sequence T807/A873 of the platelet collagen receptor integrin alpha2beta1 might be a genetic risk factor for the development of stroke in younger patients. Blood. 1999; 93:3583-6.
- Von Beckerath N, Koch W, Mehilli J et al. Glycoprotein Ia gene C807T polymorphism and risk for major adverse cardiac events within the first 30 days after coronary artery stenting. Blood. 2000; 95:3297-301.
- Morita H, Kurihara H, Imai Y et al. Lack of association between the platelet glycoprotein Ia C807T gene polymorphism and myocardial infarction in Japanese. An approach entailing melting curve analysis with specific fluorescent hybridization probes. Thromb Haemost. 2001; 85:226-30.
- Baker RI, Eikelboom J, Lofthouse EY et al. Platelet glycoprotein I b alpha Kozak polymorphism is associated with an increased risk of ischemic stroke. Blood. 2001; 98:36-40.
- Hsieh K, Funk M, Schillinger M et al. Vienna Stroke Registry. Impact of the platelet glycoprotein Ib alpha Kozak polymorphism on the risk of ischemic cerebrovascular events: a case-control study. Blood Coagul Fibrinolysis. 2004; 15:469-73.
- Yongbin N, Dayi H, Hong YW et al. Association of genetic polymorphisms in the fibrinogen and platelet glycoprotein genes with unstable angina in Chinese patients. Clin Cardiol. 2004; 27:455-8.
- Afshar-Kharghan V, Li CQ, Khoshnevis-Asl MJ et al. Kozak sequence polymorphism of the glycoprotein (GP) Ibalpha gene is a major determinant of the plasma membrane levels of the platelet GP Ib-IX-V complex. Blood. 1999; 94:186-91.
- Lopez JA, Ludwig EH, McCarthy BJ. Polymorphism of human glycoprotein Ib alpha results from a variable number of tandem repeats of a 13-amino acid sequence in the mucin-like macroglycopeptide region. Structure/function implications. J Biol Chem.1992 May; 267:10055-61.
- Fontana P, Dupont A, Gandrille S et al. Adenosine diphosphate-induced platelet aggregation is associated with P2Y12 gene sequence variations in healthy subjects. Circulation. 2003; 108:989-95.
- Fontana P, Gaussem P, Aiach M et al. P2Y12 H2 haplotype is associated with peripheral arterial disease: a case-control study.Circulation. 2003; 108:2971-3.
- Rand ML, Leung R, Packham MA. Platelet function assays. Transfus Apheresis Sci. 2003; 28:307-17.
- Yngen M, Li N, Hjemdahl P, Wallen NH. Insulin enhances platelet activation in vitro. Thromb Res. 2001; 104:85.
- Zdrojewski Z, Lizakowski S, Raszeja-Specht A et al. Influence of spontaneous platelet aggregation on progression of glomerular disease. Nephron. 2002; 92:36-42.
- Juhan I, Vague P, Buonocore M et al. Effects of insulin on erythrocyte deformability in diabetics--relationship between erythrocyte deformability and platelet aggregation. Scand J Clin Lab Invest Suppl. 1981; 156:159-64.
- Cho NH, Becker D, Dorman JS et al. Spontaneous whole blood platelet aggregation in insulin-dependent diabetes mellitus: an evaluation in an epidemiologic study. Thromb Haemost. 1989; 61:127-30.
- Mandal S, Sarode R, Dash S et al. Hyperaggregation of platelets detected by whole blood platelet aggregometry in newly diagnosed noninsulin-dependent diabetes mellitus. Am J Clin Pathol. 1993; 100:103-7.
- Golanski J, Golanski R, Chizynski K et al. Platelet hyperreactivity after coronary artery bypass grafting: the possible relevance to glycoprotein polymorphisms. A preliminary report. Platelets 2001; 12:241-7.
- Heilmann E, Hynes LA, Burstein SA et al. Fluorescein derivatization of fibrinogen for flow cytometric analysis of fibrinogen binding to platelets. Cytometry. 1994 1;17:287-93.
- Faraday N, Goldschmidt-Clermont P, Dise K et al. Quantitation of soluble fibrinogen binding to platelets by fluorescence-activated flow cytometry. J Lab Clin Med. 1994; 123:728-40.
- Aupeix K, Hugel B, Martin T, Bischoff P et al. The significance of shed membrane particles during programmed cell death in vitro, and in vivo, in HIV-1 infection. J Clin Invest. 1997; 99:1546-54.
- Mahla E, Lang T, Vicenzi MN, Werkgartner G et al. Thromboelastography for monitoring prolonged hypercoagulability after major abdominal surgery. Anesth Analg. 2001; 92:572-7.
- Pihusch R, Rank A, Gohring P et al. Platelet function rather than plasmatic coagulation explains hypercoagulable state in cholestatic liver disease. J Hepatol. 2002; 37:548-55.
- Frossard M, Fuchs I, Leitner JM et al. Platelet function predicts myocardial damage in patients with acute myocardial infarction. Circulation. 2004; 110:1392-7.
- Knobler H, Savion N, Shenkman B et al. Shear-induced platelet adhesion and aggregation on subendothelium are increased in diabetic patients. Thromb Res. 1998; 90:181-90.
- Eto K, Ochiai M, Isshiki T et al. Platelet aggregability under shear is enhanced in patients with unstable angina pectoris who developed acute myocardial infarction. Jpn Circ J. 2001; 65:279-82.
- Hekimsoy Z, Payzin B, Ornek T et al. Mean platelet volume in Type 2 diabetic patients. J Diabetes Complications. 2004; 18:173-6.
- Mimidis K, Papadopoulos V, Kotsianidis J et al. Alterations of platelet function, number and indexes during acute pancreatitis. Pancreatology. 2004; 4:22-7.
- Bath P, Algert C, Chapman N et al; PROGRESS Collaborative Group. Association of mean platelet volume with risk of stroke among 3134 individuals with history of cerebrovascular disease. Stroke. 2004; 35:622-6.
- Collins CE, Cahill MR, Newland AC et al. Platelets circulate in an activated state in inflammatory bowel disease. Gastroenterology. 1994; 106:840-5.
- Wallen NH, Held C, Rehnqvist N et al. Effects of mental and physical stress on platelet function in patients with stable angina pectoris and healthy controls. Eur Heart J. 1997; 18:807-15.
- Buyukasyk NS, Ileri M, Alper A et al. Increased blood coagulation and platelet activation in patients with infective endocarditis and embolic events. Clin Cardiol. 2004; 27:154-8.
- Goette A, Weber M, Lendeckel U et al. Effect of physical exercise on platelet activity and the von-Willebrand-factor in patients with persistent lone atrial fibrillation.J Interv Card Electrophysiol. 2004; 10:139-46.
- Born GVR. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962;194: 927-9.
- Jarvis GE. Platelet aggregation in whole blood. Impedance and particle counting methods. In: Methods in molecular biology, vol.272: Platelets and megakaryocytes, Vol. 1: Functional assays, eds. Gibbins JM, Mahaut- Smith MP. Humana Press Inc., 2004, Totowa, NJ ,77-87.
- Riess H, Braun G, Brehm G et al. Critical evaluation of platelet aggregation in whole human blood. Am J Clin Pathol. 1986; 85:50-6.
- Dyszkiewicz-Korpanty A, Frenkel EP, Sarode R. Approach to the assessment of platelet function: comparison between optical-based platelet-rich plasma and impedance -based whole blood aggregation methods. Clin Appl Thromb Hemost 2004; 10(4): in press.
- De La Cruz JP, Paez MV, Carmona JA et al. Antiplatelet effect of the anaesthetic drug propofol: influence of red blood cells and leucocytes.Br J Pharmacol. 1999; 128:1538-44.
- Klein B, Faridi A, Von Tempelhoff GF et al. A whole blood flow cytometric determination of platelet activation by unfractionated and low molecular weight heparin in vitro. Thromb Res. 2002; 108:291-6.
- Ruf A, Patscheke H. Flow cytometric detection of activated platelets: comparison of determining shape change, fibrinogen binding, and P-selectin expression. Semin Thromb Hemost. 1995; 21:146-51.
- Heilmann E, Hynes LA, Burstein SA et al. Fluorescein derivatization of fibrinogen for flow cytometric analysis of fibrinogen binding to platelets . Cytometry 1994; 17:287-293.
- Faraday N, Goldshmidt-ClermontP, Dise K et al. Quantitation of soluble fibrinogen binding to platelets by florescence- activated flow cytometry. J Lab Clin Med 1994; 123:728-40.
- Michelson AD, Rajasekhar D, Bednarek FJ et al. Platelet and platelet-derived microparticle surface factor V/Va binding in whole blood: differences between neonates and adults. Thromb Haemost. 2000; 84:689-94.
- Michelson AD, Barnard MR, Krueger LA et al. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation. 2001; 104:1533-7.
- Furman MI, Barnard MR, Krueger LA et al. Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol. 2001; 38:1002-6.
- Michelson AD, Barnard MR, Krueger LA et al. Flow cytometry. In: Platelets, ed. Michelson AD, Academic Press, Elsevier Science, 2002, San Diego, CA, USA& London, UK, 297-315.
- Gum PA, Kottke-Marchant K, Poggio ED et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol. 2001; 88:230-5.
- Gum PA, Kottke-Marchant K, Welsh PA et al. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol. 2003; 41:961-5.
- Alberts MJ, Bergman DL, Molner E et al. Antiplatelet effect of aspirin in patients with cerebrovascular disease. Stroke. 2004 ; 35:175-8.
- Gurbel PA, Bliden KP, Hiatt BL et al. Clopidogrel for coronary stenting: response variability, drug resistance, and the effect of pretreatment platelet reactivity. Circulation. 2003; 107:2908-13.
- Lau WC, Gurbel PA, Watkins PB et al. Contribution of hepatic cytochrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation. 2004; 109:166-71.
- Undas A, Brummel K, Musial J et al. Pl(A2) polymorphism of beta(3) integrins is associated with enhanced thrombin generation and impaired antithrombotic action of aspirin at the site of microvascular injury. Circulation. 2001; 104:2666-72.
- Dropinski J, Sanak M, Wegrzyn W et al. Platelet glycoprotein IIIa polymorphism and complementary actions of aspirin and clopidogrel in coronary artery disease. In: Proceedings in coronary artery disease. Ed. Monduzzi, Medimond Inc, Florence, Italy, 2003, 515-19.
- Angiolillo DJ, Fernandez-Ortiz A, Bernardo E et al. PlA polymorphism and platelet reactivity following clopidogrel loading dose in patients undergoing coronary stent implantation.Blood Coagul Fibrinolysis. 2004; 15:89-93.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol. 2003; 28:317-47.
- Frenkel EP, Bick RL. Prothrombin G20210A gene mutation, heparin cofactor II defects, primary (essential) thrombocythemia, and thrombohemorrhagic manifestations. Semin Thromb Hemost. 1999; 25:375-86.
- Frenkel EP, Mammen EF. Sticky platelet syndrome and thrombocythemia. Hematol Oncol Clin North Am. 2003; 17:63-83.
- Schafer AI. Thrombocytosis. N Engl J Med. 2004; 350:1211-9.
- Frenkel EP. The clinical spectrum of thrombocytosis and thrombocythemia. Am J Med Sci. 1991; 301:69-80.
- Vardiman JB, Harris NL. Chronic myeloproliferative diseases: Introduction, In Pathology and genetics of tumours of haematopoietic and lymphoid tissues, H.N. Jaffe ES, Stein H, Vardiman JW, Editor. 2001, IARC Press: Lyon, 17-19.
- Ruggeri M, Tosetto A, Frezzato M et al. The rate of progression to polycythemia vera or essential thrombocythemia in patients with erythrocytosis or thrombocytosis. Ann Intern Med. 2003; 139:470-5.
- Kaushansky K. Regulation of megakaryopoiesis, In Thrombosis and hemorrhage, S. AI, Editor. 2003, Williams & Wilkins: Philadelphia, USA, 120-139.
- Wang JC, Chen C, Novetsky AD et al. Blood thrombopoietin levels in clonal thrombocytosis and reactive thrombocytosis. Am J Med, 1998. 104:451-5.
- Hsu HC, Tsai WH, Jiang ML et al. Circulating levels of thrombopoietic and inflammatory cytokines in patients with clonal and reactive thrombocytosis. J Lab Clin Med, 1999; 134:392-7.
- Folman CC, Ooms M, Kuenen BB et al. The role of thrombopoietin in post-operative thrombocytosis. Br J Haematol, 2001; 114:126-33.
- Ishiguro A, Suzuki Y, Mito M et al. Elevation of serum thrombopoietin precedes thrombocytosis in acute infections. Br J Haematol. 2002; 116:612-8.
- Kaser A, Brandacher G, Steurer W et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood. 2001; 98:2720-5.
- Wolber EM, Fandrey J, Frackowski U et al. Hepatic thrombopoietin mRNA is increased in acute inflammation. Thromb Haemost. 2001; 86:1421-4.
- Espanol I, Hernandez A, Cortes M et al. Patients with thrombocytosis have normal or slightly elevated thrombopoietin levels. Haematologica. 1999; 84:312-6.
- Marta R, Goette N, Lev P et al. Increased levels of plasma interleukin-6 soluble receptor in patients with essential thrombocythemia. Haematologica. 2004; 89:657-63.
- Horikawa Y, Matsumura I, Hashimoto K et al. Markedly reduced expression of platelet c-mpl receptor in essential thrombocythemia. Blood 1997;90:4031-8.
- Li J, Xia Y, Kuter DJ. The platelet thrombopoietin receptor number and function are markedly decreased in patients with essential thrombocythaemia. Br J Haematol. 2000; 111:943-53.
- Mesa RA, Hanson CA, Li CY et al. Diagnostic and prognostic value of bone marrow angiogenesis and megakaryocyte c-Mpl expression in essential thrombocythemia. Blood. 2002; 99:4131-7.
- Teofili L, Pierconti F, Di Febo A et al. The expression pattern of c-mpl in megakaryocytes correlates with thrombotic risk in essential thrombocythemia. Blood. 2002. 100:714-7.
- Axelrad AA, Eskinazi D, Correa PN et al. Hypersensitivity of circulating progenitor cells to megakaryocyte growth and development factor (PEG-rHu MGDF) in essential thrombocythemia. Blood. 2000. 96:3310-21.
- Kuroda H, Matsunaga T, Terui T et al. Decrease of Smad4 gene expression in patients with essential thrombocythaemia may cause an escape from suppression of megakaryopoiesis by transforming growth factor-beta1. Br J Haematol. 2004. 124:211-20.
- Michiels JJ, Juvonen E. Proposal for revised diagnostic criteria of essential thrombocythemia and polycythemia vera by the Thrombocythemia Vera Study Group. Semin Thromb Hemost. 1997; 23:339-47.
- Imbert MP, Thiele R, Vardiman J et al. Essential thrombocythaemia, in pathology and genetics of tumours of haematopoietic and lymphoid tissues, Eds. Jaffe ES, Stein H, Vardiman JW, 2001, IARC Press: Lyon, 39-41.
- Laszlo J. Myeloproliferative disorders (MPD): myelofibrosis, myelosclerosis, extramedullary hematopoiesis, undifferentiated MPD, and hemorrhagic thrombocythemia. Semin Hematol. 1975;12:409-32.
- Iland HJ, Laszlo J, Peterson P et al. Essential thrombocythemia: clinical and laboratory characteristics at presentation. Trans Assoc Am Physicians.1983; 96:165-74.
- Iland HJ, Laszlo J, Case DC Jr. et al. Differentiation between essential thrombocythemia and polycythemia vera with marked thrombocytosis. Am J Hematol. 1987;25:191-201.
- Gilbert HS. Diagnosis and treatment of thrombocythemia in myeloproliferative disorders. Oncology (Huntingt). 2001;15: 989-96, 998; discussion 999-1000, 1006, 1008.
- Murphy S. Thrombocytosis and thrombocythaemia. Clin Haematol. 1983;12:89-106.
- Schafer AI. Bleeding and thrombosis in the myeloproliferative disorders. Blood. 1984;64:1-12.
- Schilling RF. Platelet millionaires. Lancet.1980; 2:372-3.
- Hoagland HC and Silverstein MN. Primary thrombocythemia in the young patient. Mayo Clin Proc. 1978;53:578-80.
- Griesshammer M, Klippel S, Strunck E et al. PRV-1 mRNA expression discriminates two types of essential thrombocythemia. Ann Hematol. 2004;83:364-70.
- Klippel S, Strunck E, Temerinac S et al. Quantification of PRV-1 mRNA distinguishes polycythemia vera from secondary erythrocytosis. Blood. 2003; 102:3569-74.
- Klippel S, Strunck E, Busse CE et al. Biochemical characterization of PRV-1, a novel hematopoietic cell surface receptor, which is overexpressed in polycythemia rubra vera. Blood. 2002; 100:2441-8.
- Cilloni D, Carturan S, Gottardi E et al. Usefulness of the quantitative assessment of PRV-1 gene expression for the diagnosis of polycythemia vera and essential thrombocythemia patients. Blood. 2004;103: 2428; author reply 2429.
- Tefferi A., T.L. Lasho, A.P. Wolanskyj et al. Neutrophil PRV-1 expression across the chronic myeloproliferative disorders and in secondary or spurious polycythemia. Blood. 2004;103: 3547-8.
- Temerinac S., S. Klippel, E. Strunck et al. Cloning of PRV-1, a novel member of the uPAR receptor superfamily, which is overexpressed in polycythemia rubra vera. Blood. 2000; 95:2569-76.
- Palmqvist L, Goerttler P, Wasslavik C et al. Comparison of methods for polycythemia rubra vera-1 mRNA quantification in whole-blood leukocytes and purified granulocytes. Clin Chem.2004; 50:644-7.
- Liu E, Jelinek J, Pastore YD et al. Discrimination of polycythemias and thrombocytoses by novel, simple, accurate clonality assays and comparison with PRV-1 expression and BFU-E response to erythropoietin. Blood. 2003; 101:3294-301.
- Florensa L, Besses C, Zamora L et al. Endogenous erythroid and megakaryocytic circulating progenitors, HUMARA clonality assay, and PRV-1 expression are useful tools for diagnosis of polycythemia vera and essential thrombocythemia. Blood. 2004; 103:2427-8.
- Mesa RA, Silverstein MN, Jacobsen SJ et al. Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: an Olmsted County Study, 1976-1995. Am J Hematol. 1999; 61:10-5.
- McIntyre KJ, Hoagland HC, Silverstein MN et al. Essential thrombocythemia in young adults. Mayo Clin Proc.1991; 66:149-54.
- Murphy S, Iland H, Rosenthal D et al. Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin Hematol. 1986; 23:177-82.
- Randi ML, Putti MC, Fabris F et al. Features of essential thrombocythaemia in childhood: a study of five children. Br J Haematol. 2000; 108:86-9.
- Van Genderen PJ and Michiels JJ. Erythromelalgia: a pathognomonic microvascular thrombotic complication in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost 1997; 23:357-63.
- Michiels JJ, Abels J, Steketee J et al. Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med, 1985; 102:466-71.
- Michiels JJ, Ten Kate FW, Vuzevski VD et al. Histopathology of erythromelalgia in thrombocythaemia. Histopathology.1984; 8:669-78.
- Griesshammer M, Bangerter M, Van Vliet HH et al. Aspirin in essential thrombocythemia: status quo and quo vadis. Semin Thromb Hemost. 1997; 23:371-7.
- Schroer K. Aspirin and platelets: the antiplatelet action of aspirin and its role in thrombosis treatment and prophylaxis. Semin Thromb Hemost.1997; 3:349-356.
- Preston FE, Emmanuel IG, Winfield DA et al. Essential thrombocythaemia and peripheral gangrene. Br Med J.1974; 3:48-52.
- Bellucci S, Janvier M, Tobelem G et al. Essential thrombocythemias. Clinical evolutionary and biological data. Cancer.1986; 8:2440-7.
- Murphy S, Peterson P, Iland H et al. Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. Semin Hematol. 1997; 34:29-39.
- Pearson TC. Primary thrombocythaemia: diagnosis and management. Br J Haematol. 1991; 78:145-8.
- Jantunen R, Juvonen E, Ikkala E et al. Essential thrombocythemiadiagnosis: causes of diagnostic evaluation and presence of positive diagnostic findings. Ann Hematol. 1998;77:101-6.
- Koudstaal PJ and Koudstaal A. Neurologic and visual symptoms in essential thrombocythemia: efficacy of low-dose aspirin. Semin Thromb Hemost. 1997;23:365-70.
- Budde U and Van Genderen PJ. Acquired von Willebrand disease in patients with high platelet counts. Semin Thromb Hemost. 1997; 23:425-31.
- Kondo T, Okabe M, Sanada M et al. Familial essential thrombocythemia associated with one-base deletion in the 5’-untranslated region of the thrombopoietin gene. Blood. 1998; 92:1091-6.
- Murphy S. Diagnostic criteria and prognosis in polycythemia vera and essential thrombocythemia. Semin Hematol. 1999;36(Suppl 2):9-13.
- Wehmeier A, Sudhoff T and Meierkord F. Relation of platelet abnormalities to thrombosis and hemorrhage in chronic myeloproliferative disorders. Semin Thromb Hemost. 1997; 23:391-402.
- Cincotta R, Higgins JR, Tippett C et al. Management of essential thrombocythaemia during pregnancy. Aust N Z J Obstet Gynaecol. 2000;40:33-7.
- Falconer J, Pineo G, Blahey W et al. Essential thrombocythemia associated with recurrent abortions and fetal growth retardation. Am J Hematol. 1987; 25:345-7.
- Mercer B, Drouin J, Jolly E et al. Primary thrombocythemia in pregnancy: a report of two cases. Am J Obstet Gynecol. 1988;159:127-8.
- Snethlage W and Ten Cate JW. Thrombocythaemia and recurrent late abortions: normal outcome of pregnancies after antiaggregatory treatment. Case report. Br J Obstet Gynaecol. 1986;93:386-8.
- Ding J, Komatsu H, Wakita A et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood. 2004; 103:4198-200.
- Cortelazzo S, Viero P, Finazzi G et al. Incidence and risk factors for thrombotic complications in a historical cohort of 100 patients with essential thrombocythemia. J Clin Oncol. 1990; 8:556-62.
- Cortelazzo S, Finazzi G, Ruggeri M et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995; 332:1132-6.
- Landolfi R, Marchioli R and Patrono C. Mechanisms of bleeding and thrombosis in myeloproliferative disorders. Thromb Haemost. 1997; 78:617-21.
- Schafer AI. Management of thrombocythemia. Curr Opin Hematol. 1996;3:341-6.
- Regev A, Stark P, Blickstein D et al. Thrombotic complications in essential thrombocythemia with relatively low platelet counts. Am J Hematol. 1997;56:168-72.
- Frenkel EP. The clinical spectrum of thrombocytosis and thrombocythemia. In: University of Texas Southwestern Medical Center Internal Medicine Grand Rounds. 1990. Dallas, TX.
- Frenkel EP. Polycythemia vera, myelofibrosis, and primary (essential) thrombocythemia. In: Medical Oncology: Basic Principles and Clinical Management, Ed. P.S. Calabresse,1993, McGraw Hill: New York, USA, 503-515.
- Frenkel EP. Myeloproliferative disorders: Polycythemia vera, thrombocythemia and myelofibrosis, in Textbook of Internal Medicine, W. Kelly, Editor. 1997, Lippincott-Raven: Philadelphia, USA, 1388-1392.
- Ravandi-Kashani F and Schafer AI. Microvascular disturbances, thrombosis, and bleeding in thrombocythemia: current concepts and perspectives. Semin Thromb Hemost. 1997;23:479-88.
- Manoharan A, Gemmell R, Brighton T et al. Thrombosis and bleeding in myeloproliferative disorders: identification of at-risk patients with whole blood platelet aggregation studies. Br J Haematol.1999;105:618-25.
- Sacher RA, Jacobson RJ and McGill M. Functional and morphological studies of platelet reactivity with vessel wall subendothelium in chronic myeloproliferative disease. Br J Haematol, 1981;49:43-52.
- Boughton BJ, Allington MJ and King A. Platelet and plasma beta thromboglobulin in myeloproliferative syndromes and secondary thrombocytosis. Br J Haematol. 1978; 40:125-32.
- Wehmeier A, Fricke S, Scharf RE et al. A prospective study of haemostatic parameters in relation to the clinical course of myeloproliferative disorders. Eur J Haematol 1990; 45:191-7.
- Wu K. Platelet hyperaggregability and thrombosis in patients with thrombocythemia. Ann Intern Med. 1978; 88:7-11.
- Bellucci S, Ignatova E, Jaillet N et al. Platelet hyperactivation in patients with essential thrombocythemia is not associated with vascular endothelial cell damage as judged by the level of plasma thrombomodulin, protein S, PAI-1, t-PA and vWF. Thromb Haemost. 1993; 70:736-42.
- Gisslinger H, Rodeghiero F, Ruggeri M et al. Homocysteine levels in polycythaemia vera and essential thrombocythaemia. Br J Haematol. 1999; 105:551-5.
- Fiedler W, Henke RP, Ergun S et al. Derivation of a new hematopoietic cell line with endothelial features from a patient with transformed myeloproliferative syndrome: a case report. Cancer. 2000; 88:344-51.
- Mesa RA, Hanson CA, Rajkumar SV et al. Evaluation and clinical correlations of bone marrow angiogenesis in myelofibrosis with myeloid metaplasia. Blood. 2000;96:3374-80.
- Verheul HM, Jorna AS, Hoekman K et al. Vascular endothelial growth factor-stimulated endothelial cells promote adhesion and activation of platelets. Blood. 2000;96:4216-21.
- Usuki K, Iki S, Endo M et al. Influence of thrombopoietin on platelet activation in myeloproliferative disorders. Br J Haematol. 1997; 97:530-7.
- Cacciola RR, Di Francesco E, Ferlito C et al. Vascular endothelial growth factor and thrombopoietin in patients with essential thrombocythemia and polycythemia vera and thrombotic complications. Acta Haematol. 2003; 110:202-3.
- Kornblihtt LI, Heller PG, Correa G et al. Associated thrombophilic defects in essential thrombocythaemia: their relationship with clinical manifestations. Thromb Res. 2003; 112:131-5.
- Barbui T and Finazzi G. Clinical parameters for determining when and when not to treat essential thrombocythemia. Semin Hematol. 1999;36 (1 Suppl 2):14-8.
- Bazzan M, Tamponi G, Schinco P et al. Thrombosis-free survival and life expectancy in 187 consecutive patients with essential thrombocythemia. Ann Hematol. 1999;78:539-43.
- Michiels JJ. Normal life expectancy and thrombosis-free survival in aspirin treated essential thrombocythemia. Clin Appl Thromb Hemost. 1999; 5:30-6.
- Rozman C, Giralt M, Feliu E et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer. 1991; 67:2658-63.
- Finazzi G, Ruggeri M, Rodeghiero F et al. Second malignancies in patients with essential thrombocythaemia treated with busulphan and hydroxyurea: long-term follow-up of a randomized clinical trial. Br J Haematol. 2000;110:577-83.
- Randi ML, Rossi C, Fabris F et al. Aspirin seems as effective as myelosuppressive agents in the prevention of rethrombosis in essential thrombocythemia. Clin Appl Thromb Hemost. 1999; 5:131-5.
- Pearson TC, Bareford D, Craig J et al. The management of “low-risk” and “intermediate-risk” patients with primary thrombocythaemia. MPD (UK) Study Group. Br J Haematol. 1999; 106:833-4.
- Ruggeri M, Finazzi G, Tosetto A et al. No treatment for low-risk thrombocythaemia: results from a prospective study. Br J Haematol. 1998; 103:772-7.
- Tefferi A. Risk-based management in essential thrombocythemia. In: The American Society of Hematology Education Program Book, Eds. GH Schechter, R Schrier, SL,1999, The American Society of Hematology: New Orleans, USA, 172-177.
- Johansson P, Ricksten A, Wennstrom L et al. Increased risk for vascular complications in PRV-1 positive patients with essential thrombocythaemia. Br J Haematol. 2003; 123:513-6.
- Millard FE, Hunter CS, Anderson M et al. Clinical manifestations of essential thrombocythemia in young adults. Am J Hematol. 1990; 33:27-31.
- Sterkers Y, Preudhomme C, Lai JL et al. Acute myeloid leukemia and myelodysplastic syndromes following essential thrombocythemia treated with hydroxyurea: high proportion of cases with 17p deletion. Blood. 1998; 91:616-22.
- De Bergsagel DE, Frenkel EP, Alfrey CP Jr. et al. Megaloblastic Erythropoiesis Induced by Hydroxyurea (Nsc-32065). Cancer Chemother Rep, 1964; 40:15-7.
- Frenkel EP, Skinner WN and Smiley JD. Studies on a Metabolic Defect Induced by Hydroxyurea (Nsc-32065). Cancer Chemother Rep.1964; 40:19-22.
- Frenkel EP and Arthur C. Induced ribotide reductive conversion defect by hydroxyurea and its relationship to megaloblastosis. Cancer Res. 1967; 27:1016-9.
- Van den Anker-Lugtenburg PJ and Sizoo W. Myelodysplastic syndrome and secondary acute leukemia after treatment of essential thrombocythemia with hydroxyurea. Am J Hematol. 1990;33:152.
- Finazzi G, Ruggeri M, Rodeghiero F et al. Efficacy and safety of long-term use of hydroxyurea in young patients with essential thrombocythemia and a high risk of thrombosis. Blood. 2003;101:3749.
- Fenaux P, Simon M, Caulier MT et al. Clinical course of essential thrombocythemia in 147 cases. Cancer. 1990; 66:549-56.
- Hehlmann R, Jahn M, Baumann B et al. Essential thrombocythemia. Clinical characteristics and course of 61 cases. Cancer. 1988; 61:2487-96.
- Chott A, Gisslinger H, Thiele J et al. Interferon-alpha-induced morphological changes of megakaryocytes: a histomorphometrical study on bone marrow biopsies in chronic myeloproliferative disorders with excessive thrombocytosis. Br J Haematol. 1990; 74:10-16.
- Elliott MA and Tefferi A. Interferon-alpha therapy in polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 1997; 23:463-72.
- Giles FJ, Singer CR, Gray AG et al. Alpha-interferon therapy for essential thrombocythaemia. Lancet.1988; 2:70-2.
- Gisslinger H, Ludwig H, Linkesch W et al. Long-term interferon therapy for thrombocytosis in myeloproliferative diseases. Lancet. 1989. 1(8639):634-7.
- Petit JJ, Callis M and Fernandez de Sevilla A. Normal pregnancy in a patient with essential thrombocythemia treated with interferon-alpha 2b. Am J Hematol. 1992; 40:80.
- Silver RT. Interferon in the treatment of myeloproliferative diseases. Semin Hematol 1990; 27(3 Suppl 4):6-14.
- Gillespie E. Anagrelide: a potent and selective inhibitor of platelet cyclic AMP phosphodiesterase enzyme activity. Biochem Pharmacol. 1988; 37:2866-8.
- Abe Andes W, Noveck RJ and Fleming JS. Inhibition of platelet production induced by an antiplatelet drug, anagrelide, in normal volunteers. Thromb Haemost. 1984; 52:325-8.
- Anagrelide Study Group. Anagrelide, a therapy for thrombocythemic states: experience in 577 patients. Am J Med. 1992; 92:69-76.
- Pescatore SL and Lindley C. Anagrelide: a novel agent for the treatment of myeloproliferative disorders. Expert Opin Pharmacother. 2000; 1:537-46.
- Silverstein MN, Petitt RM, Solberg LA Jr. et al. Anagrelide: a new drug for treating thrombocytosis. N Engl J Med. 1988;318:1292-4.
- Solberg LA Jr, Tefferi A, Oles KJ et al. The effects of anagrelide on human megakaryocytopoiesis. Br J Haematol. 1997; 99:174-80.
- Tefferi A, Silverstein MN, Petitt RM et al. Anagrelide as a new platelet-lowering agent in essential thrombocythemia: mechanism of actin, efficacy, toxicity, current indications. Semin Thromb Hemost. 1997; 23:379-83.
- Thiele J, Kvasnicka HM, Fuchs N et al. Anagrelide-induced bone marrow changes during therapy of chronic myeloproliferative disorders with thrombocytosis. an immunohistochemical and morphometric study of sequential trephine biopsies. Haematologica 2003; 88:1130-8.
- Birgegard G, Bjorkholm M, Kutti J et al. Adverse effects and benefits of two years of anagrelide treatment for thrombocythemia in chronic myeloproliferative disorders. Haematologica. 2004;89:520-527.
- Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood. 2001; 97:863-6.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC

