MENINGIOMA, UN TUMOR HUMANO BENIGNO: CAMBIOS GENETICOS EN SU INICIACION Y PROGRESION
(especial para SIIC © Derechos reservados)
Coautor
M. Cerdá Nicolás*
Departamento de Patología, Facultad de Medicina, Universidad de Valencia, España*
Recepción del artículo: 6 de diciembre, 2004
Aprobación: 0 de , 0000
 Conclusión breve
La presencia de cariotipos complejos aumenta progresivamente desde los meningiomas de grado I hasta los meningiomas de grado III.
Resumen
Los meningiomas son tumores primarios del sistema nervioso central de comportamiento generalmente benigno (grado I). Algunos de ellos tienen la capacidad de recidivar y de evolucionar a meningioma atípico (grado II) y a meningioma anaplásico (grado III). Los tumores de grado I muestran generalmente un cariotipo normal o pérdida total o parcial del cromosoma 22. Los meningiomas de grado II y III están relacionados con incremento de la hipodiploidía y la presencia de distintas alteraciones estructurales (cariotipo complejo). Presentamos una revisión sobre 100 casos de meningiomas, estudiados citogenéticamente y con técnicas de hibridación in situ con fluorescencia (FISH). Asimismo estudiamos la expresión de las fosfatasas alcalinas localizadas en el gen ALPL, en un subgrupo de meningiomas con pérdidas en 1p. De los casos con alteraciones cromosómicas, 81% presentaban la monosomía total o parcial del cromosoma 22 como única anomalía. De éstos el 71% fueron meningiomas grado I, el 29%, grado II, y ningún caso de grado III. Cariotipos complejos se encontraron en 19% de los casos, de los cuales el 13% fueron de grado I, el 27% de grado II, y el 60% de grado III. Los cromosomas más frecuentemente implicados fueron el 1, el 10, el 14 y el 22. Estos hechos significan que la presencia de cariotipos complejos aumenta progresivamente desde los meningiomas de grado I a los meningiomas de grado III. Asimismo estos cariotipos son los más habituales en los tumores recidivantes.
Palabras clave
Meningioma benigno, atípico, anaplásico, citogenética, cromosoma 1, 14 y 22
Clasificación en siicsalud
Conclusión breve
La presencia de cariotipos complejos aumenta progresivamente desde los meningiomas de grado I hasta los meningiomas de grado III.
Resumen
Los meningiomas son tumores primarios del sistema nervioso central de comportamiento generalmente benigno (grado I). Algunos de ellos tienen la capacidad de recidivar y de evolucionar a meningioma atípico (grado II) y a meningioma anaplásico (grado III). Los tumores de grado I muestran generalmente un cariotipo normal o pérdida total o parcial del cromosoma 22. Los meningiomas de grado II y III están relacionados con incremento de la hipodiploidía y la presencia de distintas alteraciones estructurales (cariotipo complejo). Presentamos una revisión sobre 100 casos de meningiomas, estudiados citogenéticamente y con técnicas de hibridación in situ con fluorescencia (FISH). Asimismo estudiamos la expresión de las fosfatasas alcalinas localizadas en el gen ALPL, en un subgrupo de meningiomas con pérdidas en 1p. De los casos con alteraciones cromosómicas, 81% presentaban la monosomía total o parcial del cromosoma 22 como única anomalía. De éstos el 71% fueron meningiomas grado I, el 29%, grado II, y ningún caso de grado III. Cariotipos complejos se encontraron en 19% de los casos, de los cuales el 13% fueron de grado I, el 27% de grado II, y el 60% de grado III. Los cromosomas más frecuentemente implicados fueron el 1, el 10, el 14 y el 22. Estos hechos significan que la presencia de cariotipos complejos aumenta progresivamente desde los meningiomas de grado I a los meningiomas de grado III. Asimismo estos cariotipos son los más habituales en los tumores recidivantes.
Palabras clave
Meningioma benigno, atípico, anaplásico, citogenética, cromosoma 1, 14 y 22
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/71437
Especialidades
 Principal: Genética Humana, Neurología, Oncología,
Principal: Genética Humana, Neurología, Oncología,
Relacionadas: Cirugía, Diagnóstico por Imágenes, Genética Humana, Geriatría, Medicina Interna, Neurocirugía, Neurología, Oncología,
Enviar correspondencia a:
Concha López Ginés. Departamento de Patología, Facultad de Medicina. Av. Blasco Ibáñez 17, 46010 Valencia, España
Patrocinio y reconocimiento
Este trabajo está financiado con una ayuda FIS PI020228, FEDER-FSE (2000-2006).
MENINGIOMA, A BENIGN HUMAN TUMOR: GENETIC CHANGES IN INITIATION AND PROGRESSION
Abstract
Meningiomas are tumors of the central nervous system generally benign (grade I), and have the capacity to progress to a higher histological grade, atypical (grade II) and malignant (grade III), which are associated with an increase in biological aggressivity and/or capacity to recur. The tumor grade I is characterized by total or partial monosomy 22, and meningiomas grade II and III by hypodiploidy and structural changes (complex karyotype). In this study, we present a review of 100 cases of meningiomas, studied by cytogenetic methods and by fluorescence in situ hybridization techniques (FISH). We have also studied the expression of alkaline phosphatases located in the ALPL gene, in a subgroup of tumors with losses in 1p. Total or partial monosomy of chromosome 22 was present in 81% of those cases having chromosomal aberrations. Of these 71% were meningiomas grade I, 29% grade II, and none were grade III. Complex karyotypes were found in 19% of cases, the 13% of which were grade I, 27% grade II, and 60% grade III. The more frequently implicated chromosomes were 1, 10, 14 and 22. These findings imply that the presence of complex karyotypes present progressively increases from grade I to grade III meningiomas. Furthermore, these karyotypes are common in recurrent tumors.
Key words
Benign atypical and malignant meningiomas, cytogenetics, chromosome 1, 14 and 22
MENINGIOMA, UN TUMOR HUMANO BENIGNO: CAMBIOS GENETICOS EN SU INICIACION Y PROGRESION
(especial para SIIC © Derechos reservados)
Artículo completo
Los meningiomas son tumores primarios del sistema nervioso central (SNC) que constituyen aproximadamente el 20% de los tumores intracraneales. Están compuestos por células neoplásicas meningoteliales derivadas de la aracnoides, de comportamiento generalmente benigno. Se presentan en adultos, con su máxima incidencia en la década de los 60 y 70 años, son muy infrecuentes en niños. Son significativamente más frecuentes en mujeres que en hombres, en una proporción 2:1.1,2
Clásicamente se diferenciaron muchos subtipos histológicos, los más frecuentes son las variantes sincitial, fibroblástica y transicional, pero la mayoría de ellas sin significación pronóstica. Una de las más importantes complicaciones en los meningiomas es su capacidad de recidivar, bien por resecciones incompletas o por agresividad biológica propia del tumor.1,3 La Organización Mundial de la Salud (OMS) considera tres grados de agresividad: grado I o meningiomas benignos, grado II o meningiomas atípicos, y grado III o meningiomas anaplásicos.4 Criterios morfológicos como el pleomorfismo nuclear, la densidad celular, nucleolos prominentes, el número de mitosis, la aparición de necrosis y la capacidad de infiltración del tejido nervioso son criterios para definir estos grados. Los grados II y III presentarían mayor capacidad de recidivar y mayor posibilidad de metastatizar.
Estos tumores presentan el mejor crecimiento en cultivos de tejidos de todos los tumores del SNC, por lo que los datos citogenéticos son los más abundantes. La pérdida total o parcial del cromosoma 22 es el primer acontecimiento citogenético en el desarrollo de meningiomas, lo cual indica que algunos de sus genes deben estar implicados en la aparición de estos tumores. Sin embargo, aparecen otras alteraciones cromosómicas tanto numéricas como estructurales constituyendo un cariotipo complejo. Entre las anomalías numéricas más importantes cabe destacar las monosomías de los cromosomas 10, 14, 18 y gonosomas, y entre las estructurales las que afectan los cromosomas 6, 7, 10, 11, 13, 14, 15 y 18,5 así como de manera esencial el cromosoma 1 en la región 1p32-1p36.6-9 Todos estos cambios secundarios se asocian con la presencia de características histopatológicas atípicas y anaplásicas así como con comportamiento clínico de mayor agresividad en la evolución del tumor. Los estudios citogenéticos en meningiomas recidivantes no son muy abundantes pero la mayoría de los casos presentan un cariotipo con diferentes anomalías además de la monosomía 22.10-12 Asimismo existe en la casuística revisada un peqeño número de meningiomas que junto con distintas alteraciones en su cariotipo conservan los dos cromosomas 22 (disomía 22).13-15 Las explicaciones que se dan a este hecho son de dos tipos: o bien que tras la pérdida del cromosoma 22 el otro se duplique quedando así este cromosoma en homocigosis y siendo por tanto compatible con la monosomía 22, o bien una explicación a nivel molecular, de manera que tendría lugar una microdeleción o una mutación en uno o varios locus del cromosoma 22, esto no tendría expresión a nivel citogenético y se podrían observar los dos cromosomas 22.
Nuestra experiencia se basa en el estudio de 100 tumores intervenidos quirúrgicamente en el Hospital Clínico Universitario de Valencia. Para establecer el diagnóstico histopatológico de las neoplasias y el grado morfológico de agresividad, se aplicaron los criterios establecidos por la OMS4 (figura 1A).
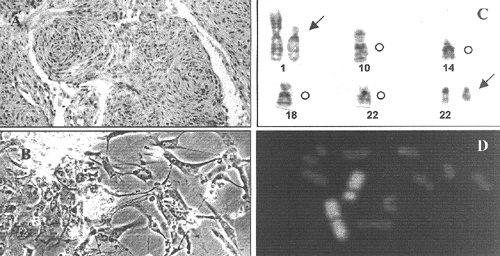
Figura 1. A) Meningioma benigno sincitial (H.E. obj. 10x). B) Meningioma atípico. Células en cultivo a las 48 horas de la siembra: las células son bipolares con nucleolos prominentes (contraste de fases 20x). C) Anomalías cromosómicas más frecuentes: deleción del cromosoma 1p, monosomía del cromosoma 10, monosomía 14, monosomía 18, monosomía 22 y monosomía parcial del cromosoma 22. D) Meningioma anaplásico. Técnicas de hibridación in situ con fluorescencia (FISH) utilizando una sonda painting para el cromosoma 1, observándose un cromosoma 1 normal y un cromosoma 1 delecionado.
El estudio citogenético se realizó sobre cultivos celulares (figura 1B) y los cariotipos se establecieron según la International System for Human Cytogenetic Nomenclature (ISCN). Estudios complementarios se realizaron con técnicas inmunohistoquímicas y con las técnicas de hibridación in situ con fluorescencia (FISH).
De los 100 pacientes, 59 eran mujeres y 41, hombres. La mayor parte de ellos (75%) tenían entre 50 y 70 años. La localización más frecuente fue la región de la hoz (29%), seguida de la convexidad (26%). Encontramos 27 tumores correspondientes a tumores recidivantes, de los cuales 4 fueron de grado I (9%), 9 de grado II (32%) y 14 de grado III (100%).
Con respecto a los resultados citogenéticos, el 30% de los casos presentaron cariotipo normal y, de ellos, 68% correspondían a meningiomas de grado I. De los casos con alteraciones cromosómicas, 81% presentaban monosomía total o parcial del cromosoma 22 como única anomalía. De éstos 71% fueron meningiomas de grado I, 29% de grado II, y ningún caso de grado III. Cariotipos complejos se encontraron en 19% de los casos, de los cuales 13% fueron de grado I, 27% de grado II, y 60% de grado III (figura 1C). Todos estos datos demostrarían que la presencia de cariotipos complejos va en aumento con respecto al grado histológico de estos tumores, desde el 24% (grado I), al 44% (grado II), al 81% (grado III).
Las anomalías numéricas más frecuentes, además de la monosomía 22, fueron las pérdidas de los cromosomas 10, 14 y 18, y los cromosomas más frecuentemente implicados en las distintas anomalías estructurales fueron: 1, 4, 7, 14 y 22; estas alteraciones fueron también estudiadas con las técnicas de FISH (figura 1D). De todas estas alteraciones se observa que la deleción de la parte distal de los brazos cortos del cromosoma 1 es la más implicada y parece estar asociada a la progresión de meningiomas. Sin embargo, tanto en la literatura como en nuestros resultados un pequeño grupo de tumores presentan pérdidas en 1p sin mostrar signos histopatológicos de anaplasia, lo que indica que en este grupo de meningiomas benignos la alteración del cromosoma 1 probablemente no es causa suficiente para su progresión.6,16-18 Otros cambios secundarios implicados en la progresión tumoral afectan frecuentemente los cromosomas 9p, 10q y 14q, especialmente este último, por lo general como pérdida del cromosoma entero.17,19,20 En nuestros casos, de los tumores de grado I con pérdidas en 1p, sólo uno presentó además pérdida del cromosoma 14; mientras que todos los meningiomas de grado II a III, excepto uno, presentaban simultáneamente pérdidas en 1p y alteraciones en el cromosoma 14.
Según los datos referidos en la bibliografía, al igual que en nuestros resultados se podrían distinguir en los meningiomas cuatro patrones citogenéticamente distintos: grupo 0, meningiomas que presentan cariotipo normal; grupo 1, tumores que presentan sólo la monosomía total o parcial del cromosoma 22 como única anomalía; grupo 2, tumores marcadamente hipodiploides con pérdidas adicionales de otros autosomas, además de la monosomía 22, y grupo 3, meningiomas con deleción de los brazos cortos del cromosoma 1, principalmente a nivel de la región 1p32-1p36, además de otras anomalías cromosómicas que incluyen o no la monosomía 22.20 Estos patrones citogenéticos están directamente relacionados con los distintos grados histopatológicos del tumor, de manera que los grupos 0 y 1 de cariotipos se presentarían con mayor frecuencia en los meningiomas benignos de grado I; mientras que los grupos 2 y 3 aparecerían en meningiomas atípicos y anaplásicos.
Con respecto a la capacidad de los meningiomas para recidivar, se observa que en relación con las características histopatológicas, este hecho tiene lugar fundamentalmente en los meningiomas de grado II y III.1,2 Este hecho también se presenta en nuestros resultados, y si además lo relacionamos con los cariotipos que presentan estos casos, se observa que 80% de los meningiomas recidivantes presentaron cariotipos complejos.
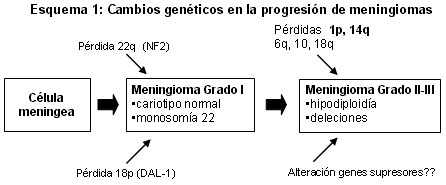
A pesar del gran conocimiento de datos citogenéticos en estos tumores, su patogénesis molecular es muy poco conocida. Mutaciones en el gen supresor de tumor NF2 (gen de la neurofibromatosis tipo II), localizado en el cromosoma 22q12.2 representarían la alteración génica más frecuente en los meningiomas. Esta alteración aparece con una frecuencia similar en los tres grados, por lo que se la consideraría un cambio en la iniciación del tumor.21,22 Recientemente otro gen –el DAL-1–, localizado en el cromosoma 18p11.3, que codifica una proteína del citoesqueleto del grupo 4.1, similar a la de NF2, se ha visto alterado también en los tres grados de meningiomas, lo que supondría un acontecimiento temprano en la tumorigénesis de estas neoplasias.23
Dado que la pérdida en el cromosoma 1p es la segunda anomalía más frecuente en estos tumores y aparece asociada con mayor agresividad biológica, se ha sugerido que genes localizados en esta región tendrían un papel relevante en la progresión tumoral. Los dos genes mejor estudiados son el p73, que codifica una proteína similar a la p53, y el gen ALPL, que codifica las fosfatasas alcalinas. Con respecto al primero, se encontraron muy pocas mutaciones, los resultados no son significativos hasta el momento.24 En el segundo gen se encontró pérdida de actividad de fosfatasa alcalina en meningiomas con pérdidas en 1p.25 Realizamos un estudio de la actividad de esta enzima en todos nuestros casos donde aparecían distintas deleciones del cromosoma 1p, estudiadas con las técnicas convencionales de citogenética, con FISH o con ambas, y encontramos que en 91% de los meningiomas con deleciones en 1p hay pérdida de actividad de fosfatasas alcalinas. Por ello este gen ALPL localizado en 1p36.1-p34, podría tener un papel importante en la progresion de estos tumores, comportándose como un gen supresor de tumor.
Otros estudios moleculares sobre genes alterados en muchos de los tumores malignos, como el p53, genes del ciclo celular, genes oncogénicos como el KRAS, HRAS o amplificaciones en CDK4 y MDM2, no dan de momento resultados significativos.22
Por todo ello concluimos que, según los datos genéticos estudiados, la presencia de monosomía 22 o de mutaciones en genes como el NF2 y el DAl-1 –o ambas características– serían acontecimientos genéticos relacionados con la iniciación del fenotipo tumoral y la aparición de meningiomas de grado I. Pérdidas cromosómicas en 1p, 14q fundamentalmente, junto con alteraciones de genes supresores localizados en estas regiones serían cambios genéticos relacionados con la progresión tumoral y la transformación de meningiomas de grado I a meningiomas de grado II-III. Igualmente estos cambios secundarios podrían representar un factor pronóstico de un subgrupo de pacientes, con alto riesgo de recidivar.
Los autores no manifiestan conflictos.
Bibliografía del artículo
- Russell D, Rubinstein L. Pathology of tumors of the nervous system. 5th ed. Williams and Wilkins, Baltimore, 1989.
- Black P. Meningiomas. Neurosurgery 1993; 32:643-657.
- Kepes J. Meningiomas: Biology, pathology and differential diagnosis. Masson Publishing, USA, 1982.
- Kleihues P, Sobin LH. World Health Organization Classification of tumors. Cancer 2000; 88:2887.
- Mitelman F, Johansson B, Mertens F. editors 2002. Mitelman database of chromosomae aberration in cancer. http://cgap.nci.nhi.gov/chromosome/Mitelman.
- Bostrom J, Muhlbauer A, Reifenberger G. Deletion mapping of the short arm of chromosome 1 identifies a common region of deletion distal to DIS496 in human meningiomas. Acta Neuropathol (Berl) 1997; 94:479-485.
- Ishino S, Hashimoto N, Fushiki S y col. Loss of material from chromosome arm 1p during malignant progression of meningioma revealed by fluorescent in situ hybridization. Cancer 1998; 83:360-366.
- Cerdá Nicolás M, López Ginés C, Pérez Bacete M y col. Histological and cytogenetic findings in benign, atipical and anaplastic human meningiomas: a study of 60 tumors. Journal of Neuro-oncology 2000; 00:1-10.
- López Ginés C, Cerdá Nicolás M, Gil Benso R y col. Loss of 1p in recurrent meningiomas: a comparative study in successive recurrences by cytogenetics and fluorescence in situ hybridization. Cancer Genet Cytogenet 2001; 125:119-124.
- López Ginés C, Cerdá Nicolás M, Barcia Salorio J y col. Cytogenetical findings of recurrent meningiomas. A study of 10 tumors. Cancer Genet Cytogenet 1995; 85:113-117.
- Cai DX, Banerjee R, Scheithauer BW y col. Chromosome 1p and 14q FISH analysis in clinicopathologic subsets of meningioma: diagnostic and prognostic implications. J Neuropathol Exp Neurol 2001; 60:628-636.
- Rey JA, Bello MJ, De Campos JM y col. Chromosomal involvement secondary to -22 in human meningiomas. Cancer Genet Cytogenet 1988; 33:275-290.
- Dumanski JP, Rouleau GA, Nordenskjoed M y col. Molecular genetic analysis of chromosome 22 in 81 cases of meningioma. Cancer Res 1990; 50:5863-5867.
- Bello MJ, Campos JM, Vaquero J y col. Chromosome 22 heterozygosity is retained in most hyperdiploid and pseudodiploid meningiomas. Cancer Genet Cytogenet 1993; 66:117-119.
- López Ginés C, Cerdá Nicolás M, Pérez Bacete M y col. Meningiomas con disomía 22: Estudio de 9 casos. Med Clin (Barc) 1998; 111:663-666.
- Lekanne-Deprez RH, Riegman PH, Van Drunen E y col. Cytogenetic, molecular genetic and pathological analyses in 126 meningiomas. J Neuropathol Exp Neurol 1995; 54(2):224-235.
- Simon M, Von Deimling A, Larson JJ y col. Allelic losses on chromosomes 14, 10 and 1 in atypical and malignant meningiomas: a genetic model of meningioma progression. Cancer Res 1995; 55:4696-4701.
- Zang KD. Meningiomas: a cytogenetic model of a complex benign human tumor, including data on 394 karyotyped cases. Cytogenet Cell Genet 2001; 93:207-220.
- Lamszus K, Kluwe L, Matschke J y col. Allelic losses at 1p, 9q, 10q, 14q, and 22q in the progression of aggressive meningiomas and undifferentiated meningeal sarcomas. Cancer Genet Cytogenet 1999; 110:103-101.
- Ketter R, Henn W, Niedermayer I y col. Predictive value of progression-associated chromosomal aberrations for the prognosis of meningiomas: a retrospective study of 198 cases. J Neurosurgery 2001; 95:601-607.
- Kross J, De Greve K, Van Tilborg A y col. Nf2 status of meningiomas is associated with tumor localization and histology. J Pathol 2001; 194:367-372.
- Lamzus K. Meningioma Pathology, Genetics and Biology. J Neuropathol 2004; 63:275-286.
- Gutman D, Donahoe J, Perry A y col. Loss of DAL-1, a protein-related tumor suppressor, is an important early event in the pathogenesis of meningiomas. Human Mol Genet 2000; 10:1495-1500.
- Lomas J, Bello MJ, Arjona D y col. Análisis of p73 gene in meningiomas with deletion at 1p. Cancer Genet Cytogenet 2001; 129:88-91.
- Niedermayer I, Feiden W, Henn W y col. Loss of alkaline phosphatase activity in meningiomas: A rapid histochemical thecnique indicating progression-associated deletion of a putative tumor supressor gene on the distal part of the short arm of chromosome 1. J Neuropatol Exp Neurol 1997; 56:879-886.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC


