



 Artículos originales> Expertos del Mundo>
Enviar correspondencia a:
Artículos originales> Expertos del Mundo>
Enviar correspondencia a: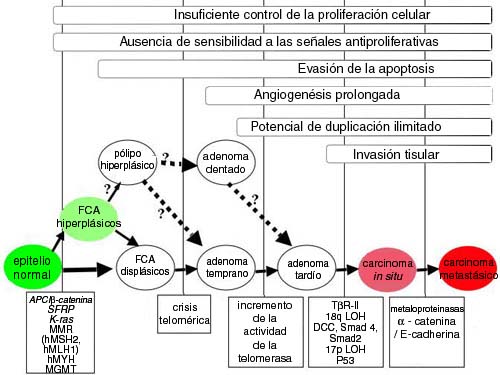
Figura 1. Progresión escalonada hacia la carcinogénesis del colon humano. Según las características morfológicas, la progresión desde la mucosa colónica normal hasta el carcinoma aparece en forma de un patrón bastante predecible. Los focos crípticos aberrantes (FCA) displásicos son los precursores más tempranos; sin embargo, datos recientes sugieren que los pólipos y los FCA hiperplásicos podrían progresar hacia FCA displásicos y adenomas. Además, diversos fenómenos genéticos se pueden observar con frecuencia en pasos similares de la vía. La inestabilidad genética, con inclusión de los defectos en CIN, MIN y BER, y la mutagénesis epigenética se presentan, de manera característica, durante el estadio inicial de la progresión y proveen un entorno permisivo para que las células adquieran mutaciones adicionales, lo que provoca la adquisición de caracteres necesarios para la transformación carcinogénica. Durante esta progresión, diversas vías de regulación y de señalización son blancos frecuentes en cualquiera de sus niveles, lo que puede alterar su expresión o función. Estas vías incluyen, entre otras, la Wnt/APC/β-catenina, BMP/TGF-β/SMAD, Src, y K-ras.La angiogénesis y el potencial de duplicación ilimitado se hallan por lo general incrementados en los pólipos tempranos a tardíos.40-42 En resumen, la transformación neoplásica es un proceso escalonado en el cual las mutaciones del ADN y la selección clonal resultan en la evolución de una célula que expresa las características comunes a todas las células cancerosas.
