CLASIFICACIONES Y SISTEMAS PRONOSTICOS EN SINDROMES MIELODISPLASICOS
(especial para SIIC © Derechos reservados)
Coautor
Dra. Irene B. Larripa*
Investigadora Principal del CONICET, Jefa del Departamento de Genética. Instituto de Investigaciones Hematológicas “Mariano R. Castex” (IIHEMA). Academia Nacional de Medicina. Ciudad Autónoma de Bueno*
Recepción del artículo: 20 de diciembre, 2004
Aprobación: 7 de febrero, 2005
 Conclusión breve
El Sistema Pronóstico Internacional (IPSS) discrimina grupos de riesgo para supervivencia y evolución leucémica teniendo en cuenta el porcentaje de blastos en médula ósea, el cariotipo y las citopenias periféricas.
Resumen
Los síndromes mielodisplásicos comprenden un grupo heterogéneo de trastornos hematológicos caracterizados por grados variables de citopenias periféricas relacionadas con una falla medular progresiva con riesgo de evolución leucémica. Dada la heterogeneidad de la patología y la dificultad de aplicar una terapéutica eficaz, se han publicado diversos sistemas de clasificación y pronóstico. El primer criterio de clasificación sistemática fue definido en 1982 por el grupo cooperativo franco-americano-británico (FAB) reconociendo entidades morfológicas: anemia refractaria (AR), AR con sideroblastos anillados (ARSA), AR con exceso de blastos (AREB), AREB en transformación (AREBt), y leucemia mielomonocítica crónica (LMMC). Aunque es la clasificación de mayor reconocimiento internacional, posee ciertas falencias pronósticas. Consecuentemente, desde 1985 se registran diferentes sistemas de predicción que toman en cuenta ciertas variables clínicas e incorporan, en 1993, el estudio citogenético. Finalmente, en 1997 se genera el Sistema Pronóstico Internacional (IPSS), el cual discrimina grupos de riesgo para supervivencia y evolución leucémica teniendo en cuenta el porcentaje de blastos en médula ósea, el cariotipo y las citopenias periféricas. El último abordaje sugerido por la Organización Mundial de la Salud, en 1999, propone un nuevo sistema de clasificación basado en hallazgos morfológicos y citogenéticos para entender esta compleja patología.
Palabras clave
síndromes mielodisplásicos, FAB, IPSS, OMS, pronóstico
Clasificación en siicsalud
Conclusión breve
El Sistema Pronóstico Internacional (IPSS) discrimina grupos de riesgo para supervivencia y evolución leucémica teniendo en cuenta el porcentaje de blastos en médula ósea, el cariotipo y las citopenias periféricas.
Resumen
Los síndromes mielodisplásicos comprenden un grupo heterogéneo de trastornos hematológicos caracterizados por grados variables de citopenias periféricas relacionadas con una falla medular progresiva con riesgo de evolución leucémica. Dada la heterogeneidad de la patología y la dificultad de aplicar una terapéutica eficaz, se han publicado diversos sistemas de clasificación y pronóstico. El primer criterio de clasificación sistemática fue definido en 1982 por el grupo cooperativo franco-americano-británico (FAB) reconociendo entidades morfológicas: anemia refractaria (AR), AR con sideroblastos anillados (ARSA), AR con exceso de blastos (AREB), AREB en transformación (AREBt), y leucemia mielomonocítica crónica (LMMC). Aunque es la clasificación de mayor reconocimiento internacional, posee ciertas falencias pronósticas. Consecuentemente, desde 1985 se registran diferentes sistemas de predicción que toman en cuenta ciertas variables clínicas e incorporan, en 1993, el estudio citogenético. Finalmente, en 1997 se genera el Sistema Pronóstico Internacional (IPSS), el cual discrimina grupos de riesgo para supervivencia y evolución leucémica teniendo en cuenta el porcentaje de blastos en médula ósea, el cariotipo y las citopenias periféricas. El último abordaje sugerido por la Organización Mundial de la Salud, en 1999, propone un nuevo sistema de clasificación basado en hallazgos morfológicos y citogenéticos para entender esta compleja patología.
Palabras clave
síndromes mielodisplásicos, FAB, IPSS, OMS, pronóstico
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/71509
Especialidades
 Principal: Hematología,
Principal: Hematología,
Relacionadas: Diagnóstico por Laboratorio, Medicina Interna,
Enviar correspondencia a:
Carolina Belli. Pacheco de Melo 3081 CP 1425, Ciudad Autónoma de Buenos Aires, Argentina.
Patrocinio y reconocimiento
Agradecimientos: Se agradece al Lic. Francisco Lastiri por el asesoramiento estadístico. Este trabajo fue subsidiado por la Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), el Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) y la Fundación Roemmers.
CLASSIFICATION AND PROGNOSTIC SYSTEMS IN MYELODYSPLASTIC SYNDROMES
Abstract
Myelodysplastic syndrome (MDS) comprises a group of heterogeneous hematological disorders with risk of leukaemic evolution (LE), characterized by varied degrees of peripheral cytopenias related to a progressive bone marrow (BM) failure. Due to the heterogeneity of this pathology and the difficulty to make decisions regarding therapy, different classification and prognostic systems have been developed. The French-American-British cooperative group defined the first criterion for a systematic classification in 1982 that recognized morphologic entities: refractory anemia (RA), RA with ringed sideroblasts (RARS), RA with excess of blast (RAEB), RAEB in transformation (RAEBt) and chronic myelomonocytic leukemia (CMML). Although the FAB is the classification with major agreement, it has some prognostic failures. Therefore, since 1985 different instruments-scoring systems for prognosis were developed taking into account diverse clinical parameters including the cytogenetic analysis in 1993. In order to standardize prognostic features in MDS the International Scoring System (IPSS) was generated in 1997. This score defined risk groups for survival and LE on the basis of percentage of BM myeloblasts, cytogenetic abnormalities and number of cytopenias. Finally in 1999, the World Health Organization proposed a new classification system based on morphology and cytogenetic findings making another intent to sort out the heterogeneity of this pathology.
Key words
Myelodysplastic syndromes, FAB, IPSS, WHO, prognostic
CLASIFICACIONES Y SISTEMAS PRONOSTICOS EN SINDROMES MIELODISPLASICOS
(especial para SIIC © Derechos reservados)
Artículo completo
Introducción
Los síndromes mielodisplásicos (SMD), también denominados síndromes dismielopoyéticos o síndromes preleucémicos,1 comprenden un grupo heterogéneo de alteraciones hematológicas caracterizadas por hematopoyesis inefectiva que resulta clínicamente en la presencia de citopenias en sangre periférica (SP) con una médula ósea (MO) normocelular a hipercelular con alteraciones displásicas. Los SMD pueden aparecer de novo o secundariamente (SMDs) al uso de agentes antineoplásicos o por contacto con químicos; estos últimos conforman alrededor del 10%. Los SMD se producen principalmente en ancianos, con un promedio de edad al momento del diagnóstico de 60-75 años, y menos del 10% de los pacientes son menores de 50 años. Se presentan con una incidencia de 3/100 000 que aumenta a 20/100 000 en personas mayores de 70 años.2
Los SMD representan un modelo atractivo para el estudio de la aparición y progresión desde un trastorno relativamente benigno hacia una neoplasia sumamente maligna. Aproximadamente 21% a 39%3-7 de los pacientes evolucionan a leucemia mieloide aguda (LMA) con una escasa respuesta a la quimioterapia. Otro 30% fallece por complicaciones vinculadas a sus citopenias (infecciones y hemorragias), y el resto, por causas no relacionadas.
Los mecanismos precisos que determinan la iniciación de los SMD son, en la actualidad, desconocidos. Podrían incluir mutaciones en el ADN nuclear o mitocondrial,8 y una reparación defectuosa del ADN que puede resultar en una inestabilidad genómica de las células hematopoyéticas troncales. Ciertas anormalidades inmunológicas8 también podrían contribuir al proceso de iniciación, estos eventos podrían ser clínicamente silentes. Un segundo paso, aun no definido, podría conferir ventajas de crecimiento al clon neoplásico, el cual se expandiría resultando en una hematopoyesis clonal que se ve reflejada en los hallazgos morfológico-clínicos de los SMD. La proliferación celular inefectiva se acompaña de una muerte celular extensiva por apoptosis de los precursores mieloides. Finalmente, posteriores daños genéticos podrían promover la evolución a LMA, con incremento en la proliferación celular y disminución de los niveles de apoptosis.6
Los SMD fueron descritos, por primera vez en 1949 como una forma subaguda de la LMA.9,10 Posteriormente fueron definidos por el Grupo Cooperativo Franco-Americano-Británico (FAB)11 de acuerdo con sus características morfológicas, la presencia de displasia en al menos dos líneas celulares y el porcentaje de blastos en MO en 5 entidades: anemia refractaria (AR), AR con sideroblastos en anillo (ARSA), AR con exceso de blastos (AREB), AREB en transformación (AREBt) y leucemia mielomonocítica crónica (LMMC). El recuento de blastos en MO mayor a 30%12 establece el diagnóstico diferencial con respecto a la LMA.
Aunque son indiscutibles los méritos de la clasificación FAB, ésta posee ciertas limitaciones en cuanto a la distinción de grupos de riesgo y a la posibilidad de predecir el comportamiento clínico de los pacientes de manera individual. Por lo tanto, con el fin de solucionar ciertas falencias, la Sociedad Europea de Hematopatólogos y la Sociedad de Hematopatología desarrollaron la nueva sistematización de la Organización Mundial de la Salud (OMS) para los SMD.2,13 Esta última se basa en la combinación de hallazgos morfológicos, inmunofenotipo, anormalidades citogenéticas y hallazgos clínicos. La OMS subdivide la AR y la ARSA en AR y ARSA simples, cuando la displasia se halla sólo en el linaje eritroide. Asimismo, se denominan citopenias refractarias con displasia multilineal (CRDM) con sideroblastos anillados o sin ellos cuando se le suma displasia de otros linajes. El subtipo síndrome 5q- se caracteriza por hallazgos displásicos en linaje eritroide, trombocitosis e hiperplasia de micromegacariocitos hipolobulados. La AREB queda subdividida, según tenga menos o más de 10% de blastos en MO, AREB-I y AREB-II. El subgrupo de SMD no clasificables incluye los pacientes que poseen citopenias refractarias con disgranulopoyesis o dismegacaripoyesis, SMD hipoplásicos, SMD con mielofibrosis, MDS hipercelular y los SMDs. Los pacientes con LMMC que poseen recuentos de GB > 13 000/μl forman parte de un nuevo grupo denominado enfermedades mielodisplásicas/mieloproliferativas y, los que poseen un recuento menor, son redistribuidos en las restantes categorías. Al mismo tiempo, elimina la AREBt fijando el límite de 20% de blastos en MO para establecer diagnóstico de LMA. Asimismo, los pacientes que presenten alteraciones citogenéticas típicas de subtipos leucémicos deben ser excluidos de los SMD y diagnosticados como LMA.
Sistemas pronósticos
La marcada heterogeneidad de los SMD se traslada en que la evolución natural de los pacientes varíe desde un curso indolente durante varios años hasta una rápida evolución a LMA, con corta supervivencia. La citada variabilidad algunas veces no puede ser explicada por el subtipo morfológico. Por ende, a partir de 1985 se desarrollaron diversos sistemas con el fin de establecer una mejor opción terapéutica.
Hasta hace muy poco tiempo el tratamiento de rutina era sólo de soporte. En la actualidad varía desde un manejo sintomatológico, que trata de mejorar el cuadro hematológico, hasta intentos de cambiar la historia natural de la enfermedad, que en comparación con la LMA de novo tiene escasa respuesta a los quimioterápicos tradicionales, menor índice de remisión completa y alto índice de recaída. Los tratamientos de baja intensidad son de elección en pacientes de bajo riesgo, y los de alta intensidad para los de alto riesgo. Sin embargo, para pacientes con mal estado general o edad avanzada sólo son aplicables los tratamientos de soporte o las terapias de baja intensidad.8 Los tratamientos que intentan cambiar el curso de la enfermedad se encuentran asociados con una alta morbimortalidad14 y sólo pueden ser aplicados a pacientes jóvenes con buen estado general.8
Los distintos sistemas de predicción toman en cuenta variables pronósticas como la presencia de citopenias en SP, la proporción de blastos en MO, la edad y el nivel plasmático de LDH. Recién en 199315 se incorpora la citogenética como variable predictiva.
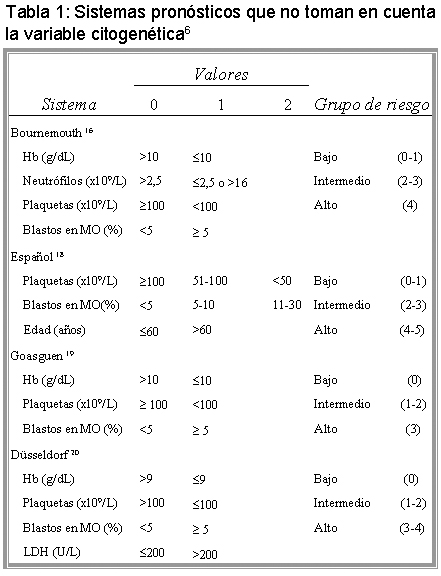
Sistemas pronósticos que no toman en cuenta la variable citogenética
El sistema Bournemouth16 fue el primer sistema pronóstico desarrollado. Posteriormente fue modificado, incluyendo la leucocitosis como variable pronóstica, con el fin de dar mayor poder discriminativo en pacientes con LMMC.17 El sistema Español18 propone entre sus variables pronósticas la edad. Su inclusión es controversial: el pronóstico ofrecido refleja parcialmente una característica propia de la población analizada y puede ser problemático para la selección de un determinado tratamiento. Es decir, los pacientes de mayor edad poseen un puntaje más alto, aunque toleran peor el tratamiento con quimioterapia que los pacientes de menor edad. Otros sistemas publicados son el Goasguen19 y el Düsseldorf.20 Este último incluye entre sus variables pronósticas el valor de la LDH. La cual califica a este sistema para el asesoramiento certero de los pacientes con LMMC, cuyos pronósticos se veían favorecidos cuando eran analizados con otros sistemas.
Sistemas pronósticos que toman en cuenta la variable citogenética
El sistema Lille15,21 fue el primero en reconocer el valor pronóstico independiente del estudio citogenético e incorporarlo en un sistema de predicción. Este sistema considera de mal pronóstico la presencia de alteraciones citogenéticas complejas y deja de lado la presencia de otras alteraciones invariablemente asociadas a un pronóstico adverso.

Con el fin de consensuar las variables pronósticas en estos síndromes se realizó un estudio cooperativo internacional cuyo resultado definió el Sistema Pronóstico Internacional (International Prognostic Scoring System: IPSS) en 1997.22 El IPSS determina que las variables de mayor impacto pronóstico son el porcentaje de blastos en MO, el análisis citogenético y el número de citopenias periféricas, con lo que quedan discriminados 4 grupos de riesgo para supervivencia y para supervivencia libre de evolución leucémica (SLL) denominados: Bajo, Intermedio 1 (Int-1), Intermedio 2 (Int-2) y Alto.
Experiencia argentina
El objetivo del trabajo científico previamente publicado por nuestro grupo7 fue evaluar la aplicación del IPSS22 en población argentina, analizar el valor pronóstico de sus variables y determinar si dicho sistema permite identificar subgrupos pronósticos de riesgo dentro de los subtipos FAB.11
Se analizaron 234 pacientes con SMD primario provenientes de instituciones argentinas con una edad promedio de 64 años (17-90), 127 pacientes eran de sexo masculino y 107 de sexo femenino. La media de seguimiento fue de 28 meses (1-196), desde 1984 hasta 2000. De los pacientes analizados, 54 (23%) evolucionaron a leucemia y 99 (42%) fallecieron por causas relacionadas (hemorragias e infecciones), incluyendo 40 pacientes que habían evolucionado previamente a LMA.
Los 234 pacientes estudiados fueron distribuidos según la Clasificación FAB11 al momento del diagnóstico y, al analizar la supervivencia y la SLL, éstas fueron significativamente diferentes entre los distintos subtipos (Tabla 3).

Asimismo, los pacientes fueron distribuidos de acuerdo con los puntos de corte estipulados por el IPSS para el porcentaje de blastos en MO y para el número de citopenias.22 Cada una de las variables mostró diferencias significativas tanto para supervivencia como para SLL (Tabla 3).
Los resultados citogenéticos (cultivos de MO de corto término, sin estimulación mitogénica) obtenidos al momento del diagnóstico mostraron que, de los 234 pacientes, 36 (15%) no presentaban material apto para el análisis citogenético y, de los 198 pacientes restantes, 82 (41%) tenían cariotipo anormal. Las anormalidades observadas de mayor frecuencia fueron: -7/del(7q) (9 pacientes), +8 (6 pacientes), del(5q) (5 pacientes), del(20q) (4 pacientes) y del(12p) (4 pacientes). Las anormalidades de menor frecuencia incluyeron del(6q) (2 pacientes), i(17q) (2 pacientes), +13 (2 pacientes). Además, 22 pacientes mostraron cariotipos complejos.
Asimismo, los hallazgos cromosómicos fueron subdivididos de acuerdo con los subgrupos de riesgo citogenético22 definidos por el IPSS (Tabla 2) y mostraron diferencias significativas con respecto a supervivencia y SLL (Tabla 3). Además, al analizar los resultados se observa que la proporción de cariotipos normales decrece a medida que aumenta el riesgo del grupo pronóstico por IPSS, es decir, de un 94% de los casos con cariotipo normal presentes en el grupo de riesgo Bajo disminuye a 17% en el grupo de riesgo Alto. Estos datos muestran la importancia del parámetro citogenético además del porcentaje de blastos y el número de citopenias.
Los 198 pacientes estudiados fueron agrupados según el IPSS22 en los respectivos grupos de riesgo (Tabla 2), los cuales mostraron diferencias significativas en cuanto a supervivencia y SLL (Tabla 3, Figura 1).

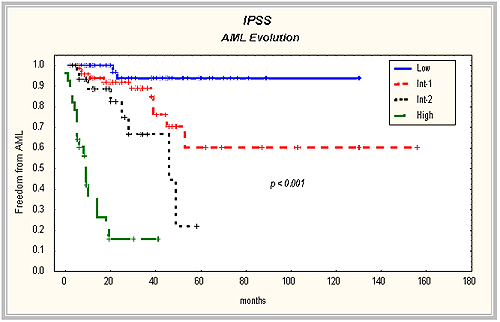
Figura 1. Curvas de supervivencia y SLL de los pacientes con SMD de acuerdo con su grupo de riesgo por el IPSS (curvas utilizando el método del límite del producto de Kaplan-Meier y el Log-Rank test).
Asimismo, los pacientes clasificados según FAB fueron agrupados de acuerdo con su grupo de riesgo IPSS, lo cual permitió individualizar pacientes con peor pronóstico dentro de los subtipos AR (5%), ARSA (15%) y AREB (19%) (Tabla 4). La adición del número de citopenias a un grupo citogenético adverso no sólo incrementa el riesgo de evolución a LMA, sino que también predice menor supervivencia, por ende, estos pacientes mostraron un riesgo mayor al previsto por la clasificación FAB.

Discusión
En el trabajo publicado previamente7 evaluamos la clasificación FAB, el IPSS y sus variables en un grupo numeroso de pacientes con diagnóstico de SMD primario, procedentes de diferentes servicios de hematología de la República Argentina. Las variables analizadas fueron predictivas tanto para supervivencia como para SLL.
El porcentaje de blastos en MO es uno de los parámetros con mayor valor pronóstico tanto en términos de supervivencia como de riesgo de evolución a LMA. El IPSS incluye un nuevo corte entre 5%-20% previamente considerados por la clasificación FAB.11 Nuestros resultados muestran que no se observan diferencias significativas ni para supervivencia ni para SLL entre 5%-10% y 11%-20%. Sin embargo, cuando se suman los grupos de riesgo citogenético y el número de citopenias, su poder discriminativo aumenta, pudiendo obtener diferencias significativas entre los cuatro grupos de riesgo según el IPSS.
El número de citopenias, determinado por la gravedad de los niveles de Hb, recuentos de plaquetas y neutrófilos, se encuentra altamente asociado con una menor supervivencia y tiene relación directa con peor pronóstico.15-21 Este hecho se traduce en un 30% de pacientes que fallecen por causas relacionadas a las mismas: hemorragias e infecciones. En nuestra serie, en particular, 59/99 fallecieron por complicaciones relacionadas con sus estados citopénicos. Además, la presencia de pancitopenia usualmente refleja la existencia de displasia multilineal, asociada también con peor pronóstico.23
La frecuencia de alteraciones cromosómicas clonales en los SMD de novo varía entre 19% y 50%,5,15,22,24 aumentando hasta un 80% en los SMDs. Y, aunque los SMD no se encuentran asociados a ninguna alteración cromosómica en particular, existe un predomino de pérdidas cromosómicas totales o parciales, una incidencia alta de ganancias y una muy baja de translocaciones. Las alteraciones son características de todo el grupo y no están asociadas a un subgrupo en particular,25-27 aunque la frecuencia se incrementa a medida que aumenta el riesgo según FAB.21 Además, pudimos observar que la proporción de resultados citogenéticos normales decrece a medida que aumenta el riesgo según el IPSS.7
Nuestros datos coinciden con los de la literatura al mostrar que la variable citogenética analizada de manera independiente posee un importante valor pronóstico tanto para supervivencia como para evolución a LMA.22,28 Las alteraciones de mayor riesgo de evolución leucémica son la –7/del(7q) y los cariotipos complejos. Mientras que los casos sin anomalías citogenéticas observables o la presencia del marcador 5q- constituyen un grupo de buen pronóstico con larga supervivencia y un riesgo disminuido de evolución neoplásica. Si analizamos las alteraciones encontradas de manera individual podemos decir que la del(5q) se asocia con buen pronóstico cuando se encuentra como única alteración, sin exceso de blastos y en pacientes de sexo femenino.7,22,28-30 Aunque sigue siendo discutido, la del(20q) aislada se asociaría con pronóstico favorable15,22,28 y la trisomía 8 con riesgo intermedio.22 Sin embargo, algunos autores las consideran con un mayor riesgo.31,32 En nuestra serie sólo 1/4 con del(20q) y 1/6 con trisomía 8 evolucionaron a LMA. Por dicho motivo, sugerimos la necesidad de un estudio que incorpore mayor número de pacientes a fin de poder determinar el impacto pronóstico de estas aberraciones. La pérdida del cromosoma Y ha sido descrita previamente en pacientes hematológicos de avanzada edad, aunque también ha sido encontrada en MO de dadores normales mayores. Por lo tanto, la presencia de dicha anormalidad no excluye la presencia de una patología clonal y una vez que el diagnóstico es confirmado mediante otros hallazgos clínicos esta alteración se encontraría asociada con pronóstico favorable.22
En nuestra población se observó que 22 (11%) pacientes presentaron cariotipos complejos, lo cual se corresponde con una frecuencia menor al 15% informada en los SMD de novo, muy inferior al 50% hallada en los SMDs.21 Estos, junto a los pacientes con –7/del(7q), mostraron escasa supervivencia y menor SLL, coincidiendo con lo previamente publicado por otros autores.22,28
La aplicación del IPSS en nuestra población de SMD permitió discriminar estadísticamente los 4 grupos de riesgo tanto para supervivencia como para SLL y fue útil para individualizar pacientes de peor pronóstico dentro de AR, ARSA y AREB. Este hecho se debe a que la adición del número de citopenias a un resultado citogenético adverso no sólo incrementa el riesgo de evolución a LMA, sino que también predice menor supervivencia.22 Con respecto al subtipo AREBt, el IPSS no aportó mayor información debido a que estos pacientes tienen mal pronóstico uniforme relacionado directamente con su alto recuento de blastos en MO. Al observar los pacientes con LMMC, el IPSS no fue predictivo debido a que a la mayoría (92%) se le adjudicó un riesgo Bajo e Int-1. La LMMC presenta características propias que pertenecen, en parte, a los SMD y en parte a los SMP, por ende, la nueva clasificación de la OMS los excluye de los SMD. Los factores pronósticos son: el número de leucocitos y de monocitos, la presencia de precursores inmaduros en SP y en MO, presencia de esplenomegalia, nivel de LDH y la presencia de alteraciones citogenéticas.2,6,13,28
La presente serie es la más grande informada en Sudamérica y es importante destacar que los resultados coinciden con lo publicado internacionalmente.4,22,31-33
Los autores no manifiestan “conflictos de interés”.
Bibliografía del artículo
- Heaney ML, Golde GW. Myelodysplasia. NEJM 1999; 340:1649-60.
- Jaffe ES, Harris NL, Stein H y col. (EDs): World Health Organization Classification of Tumours. Pathology and Genetics of tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon 2001.
- Van der Weide M, Sizoo W, Nauta J y col. Myelodysplastic syndromes: analysis of clinical and prognostic features in 96 patients. Eur J Haematol 1988; 41:115-22.
- Horiike S, Tniwaki M, Misawa S y col. Chromosome abnormalities and karyotypic evolution in 83 patients with myelodysplastic syndrome and predictive value for prognosis. Cancer 1988; 62:1129-38.
- Parlier V, Van Melle G, Beris PH y col. Prediction of 18-month survival in patients with primary myelodysplastic syndrome. A regression model and scoring system based on the combination of chromosome findings and Bournemouth score. Cancer Genet Cytogenet 1995; 81:158-65.
- Sanz GF, Sanz MA, Vallespi T. Etiopathogenesis, prognosis and therapy of myelodysplastic syndromes. Hematol Cell Ther 1997; 39:277-94.
- Belli C, Acevedo S, Bengió R y col. Detection of risk groups in myelodysplastic syndrome. A multicenter study. Haematologica 2002; 87:9-16.
- Greenberg PL, Young NL, Gatterman N. Myelodysplastic syndromes. Hematology (Am Soc Hematol Educ Program) 2002; 136-61.
- Mallarmé PJ. Les deébuts hématologiques des leucosis malignes. Sang 1949; 20:429-33.
- Hamilton-Patterson JL. Preleukaemic anaemia. Acta Haematol 1949; 2:309-16.
- Bennett JM, Catovsky D, Daniel MT y col. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 1982; 51:189-99.
- Bennett JM, Catovsky D, Daniel MT y col. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British cooperative group. Ann Intern Med 1985; 103:626-9.
- Lee Harris N, Jaffe ES, Diebold J y col. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissue: report of the Clinical Advisory Committee Meeting-Airlie House, Virginia, November 1997. J Clin Oncol 1999; 17:3835-49.
- Anderson J, Appelbaum F, Fisher L y col. Allogenic bone marrow transplantation for 93 patients with myelodysplastic síndromes. Blood 1993; 82:677-81.
- Morel P, Hebbar M, Lai JL y col. Cytogenetic analysis has strong independent prognostic value in the novo myelodysplastic syndromes and can be incorporated in a new scoring system: a report on 408 cases. Leukemia 1993; 7:1315-23.
- Mufti GJ, Stevens JR, Oscier DG y col. Myelodysplastic syndromes: a scoring system with prognostic significance. Br J Haematol 1985; 59:425-33.
- Worsley A, Oscier DG, Stevens J y col. Prognostic features of chronic myelomonocytic leukaemia: a modified Bournemouth scores gives the best prediction of survival. Br J Haematol 1988; 68:17-21.
- Sanz GF, Sanz MA, Vallespi T y col. Two regression models and a scoring system for predicting survival and planning treatment in myelodysplastic síndromes: a multivariate analysis of prognostic factors in 370 patients. Blood 1989; 74:395-408.
- Goasguen JE, Garand R, Bizet M y col. Prognostic factors of myelodysplastic syndromes. A simplified 3-D scoring system. Leuk Res 1990; 14:255-62.
- Aul C, Gatermann N, Heyll A y col. Primary myelodysplastic syndromes: analysis of prognostic factors in 235 patients and proposal for an improved scoring system. Leukemia 1992; 6:52-9
- Fenaux P, Morel P, Lai JL. Cytogenetic of myelodysplastic syndromes. Semin Hematol 1996; 33:127-38.
- Greenberg P, Cox C, LeBeau MM y col. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89:2079-88. (erratum, Blood 1998; 91:1100)
- Rosati S, Mick R, Xu F y col. Refractory cytopenia with multilineage dysplasia: further characterization of an “Unclassifiable” myelodysplastic syndrome. Leukemia 1996; 10:20-6.
- Heim S. Cytogenetic findings in primary and secondary MDS. Leuk Res 1992; 16:43-6.
- Sandberg AA. Chromosome abnormalities in human cancer and leukemia. Mutat Res 1991; 247:231-40.
- Le Beau MM. Fluorescence in situ hybridization in cancer diagnosis. Important Adv Oncol 1993; 29-45.
- Hiem S, Mitelmann F. Cytogenetic analysis in the diagnosis of acute leukemia. Cancer 1992; 15:1701-9.
- Greenberg PL. Risk factors and their relationship to prognosis in myelodysplastic syndromes. Leuk Res 1998; 22 (Suppl 1):S3-6.
- Larripa I, Acevedo S, Palau V y col. Leukaemic transformation in patients with 5q- and additional abnormalities. Haematologica 1991; 76:363-7.
- Van der Berghe H, Vermaeln K, Mecucci C y col. The 5q- anomaly. Cancer Genet Cytogenet 1985; 17:189-255.
- de Souza Fernández T, Ornellas MD, Otero de Carvalho L y col. Chromosomal alterations associated with evolution from myelodysplastic syndrome to acute myeloid leukemia. Leuk Res 2000; 24:839-48.
- Jotterand M, Parlier V. Diagnostic significance of cytogenetics in adult primary myelodysplastic syndromes. Leuk Lymphoma 1996; 23:253-66.
- Benitez J, Carbonell F, Sanchez Fayos J y col. Karyotipic evolution in patients with myelodysplastic syndromes. Cancer Genet Cytogenet 1985; 16:157-67.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC



