Bibliografía del artículo
Bibliografía del artículo
1. Silberstein SD. Migraine pathophysiology and its clinical implications. Cephalalgia 24(Suppl 2):2-7, 2004.
2. Buzzi MG, Moskowitz MA.The trigemino-vascular system and migraine. Pathol Biol (Paris) 40(4):313-7, 1992.
3. May A, Goadsby PJ. The trigeminovascular system in humans: pathophysiologic implications for primary headache syndromes of the neural influences on the cerebral circulation. J Cereb Blood Flow Metab 19(2):115-27, 1999.
4. Williamson DJ, Hargreaves RJ. Neurogenic inflammation in the context of migraine. Microsc Res Tech 53(3):167-78, 2001.
5. Geppetti P, Capone JG, Trevisani M, Nicoletti P, Zagli G, Tola MR. CGRP and migraine: neurogenic inflammation revisited. J Headache Pain 6(2):61-70, 2005.
6. Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 28(2):183-7, 1990.
7. Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 33(1):48-56, 1193.
8. Goadsby P. Neuropeptides and migraine: a useful biological marker? Cephalalgia 15(5):333-4, 1995.
9. Gallai V, Sarchielli P, Floridi A, et al. Vasoactive peptide levels in the plasma of young migraine patients with and without aura assessed both interictally and ictally. Cephalalgia 15(5):384-90, 1995.
10. Sarchielli P, Alberti A, Codini M, Floridi A, Gallai V. Nitric oxide metabolites, prostaglandins and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. Cephalalgia 20(10):907-18, 2000.
11. Ashina M, Bendtsen L, Jensen R, Schifter S, Olesen J. Evidence for increased plasma levels of calcitonin gene-related peptide in migraine outside of attacks. Pain 86(1-2):133-8, 2000.
12. Tvedskov JF, Lipka K, Ashina M, Iversen HK, Schifter S, Olesen J. No increase of calcitonin gene-related peptide in jugular blood during migraine. Ann Neurol 58(4):561-8, 2005.
13. Edvinsson L. New therapeutic target in primary headaches - blocking the CGRP receptor. Expert Opin Ther Targets 7(3):377-83, 2003.
14. Edvinsson L. Clinical data on the CGRP antagonist BIBN4096BS for treatment of migraine attacks. CNS Drug Rev 11(1):69-76, 2005.
15. Sarchielli P, Pini LA, Zanchin G, et al. Clinical-biochemical correlates of migraine attacks in rizatriptan responders and non-responders. Cephalalgia 26(3):257-65, 2006.
16. Barbanti P, Fabbrini G, Pesare M, Vanacore N, Cerbo R. Unilateral cranial autonomic symptoms in migraine. Cephalalgia 22(4):256-9, 2002.
17 Barbanti P, Fabbrini G, Vanacore N, Pesare M, Buzzi MG. Sumatriptan in migraine with unilateral cranial autonomic symptoms: an open study. Headache 43(4):400-3, 2003.
18. Thomsen LL, Olesen J. Nitric oxide in primary headaches. Curr Opin Neurol 14(3):315-21, 2001.
19. Akerman S, Williamson DJ, Kaube H, Goadsby PJ. Nitric oxide synthase inhibitors can antagonize neurogenic and calcitonin gene-related peptide induced dilation of dural meningeal vessels. Br J Pharmacol 137(1):62-8, 2002.
20. Bellamy J, Bowen EJ, Russo AF, Durham PL. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci 23(8):2057-66, 2006.
21. Iversen HK. Experimental headache in humans. Cephalalgia 15(4):281-7, 1995.
22. Thomsen LL. Investigations into the role of nitric oxide and the large intracranial arteries in migraine headache. Cephalalgia 17(8):873-95, 1997.
23. Juhasz G, Zsombok T, Modos EA, et al. NO-induced migraine attack: strong increase in plasma calcitonin gene-related peptide (CGRP) concentration and negative correlation with platelet serotonin release. Pain 106(3):461-70, 2003.
24. D'Amico D, Ferraris A, Leone M, et al. Increased plasma nitrites in migraine and cluster headache patients in interictal period: basal hyperactivity of L-arginine-NO pathway? Cephalalgia 22(1):33-6, 2002.
25. Shukla R, Barthwal MK, Srivastava N, et al. Blood nitrite levels in patients with migraine during headache-free period. Headache 41(5):475-81, 2001.
26. Lassen LH, Ashina M, Christiansen I, Ulrich V, Olesen J. Nitric oxide synthase inhibition in migraine. Lancet 349(9049):401-2, 1997.
27. Krymchantowski AV, Barbosa JS. Rizatriptan combined with rofecoxib vs. rizatriptan for the acute treatment of migraine: an open label pilot study: Cephalalgia 22(4):309-12, 2002.
28. Krymchantowski AV, Bigal ME. Rizatriptan versus rizatriptan plus rofecoxib versus rizatriptan plus tolfenamic acid in the acute treatment of migraine. BMC Neurol 4:10, 2004.
29. Saper J, Dahlof C, So Y, et al; Rofecoxib Protocol 162 Study Group. Rofecoxib in the acute treatment of migraine: a randomized controlled clinical trial. Headache 46(2):264-75, 2006.
30. Krymchantowski AV, Bigal ME. Rofecoxib in migraine. Expert Rev Neurother 5(1):55-61, 2005.
31. Schuh-Hofer S, Tayefeh M, Reuter U, Dirnagl U, Arnold G. Effects of parecoxib on plasma protein extravasation and c-fos expression in the rat. Headache 46(2):276-85, 2006.
32. Durham PL, Russo AF. Stimulation of the calcitonin gene-related peptide enhancer by mitogen-activated protein kinases and repression by an antimigraine drug in trigeminal ganglia neurons. J Neurosci 23(3):807-15, 2003.
33. Edvinsson L, Uddman R. Neurobiology in primary headaches. Brain Res Brain Res Rev 48(3):438-56, 2005.
34. Reuter U, Bolay H, Jansen-Olesen I, et al. Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain 124(Pt 12):2490-2502, 2001.
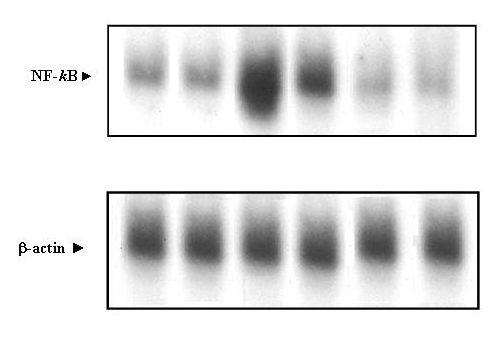
35. Reuter U, Chiarugi A, Bolay H, Moskowitz MA. Nuclear factor-kappaB as a molecular target for migraine therapy. Ann Neurol 51(4):507-16, 2002.
36. Sarchielli P, Floridi A, Mancini M, et al. NF-kappaB activity and iNOS expression in monocytes from internal jugular blood of migraine without aura patients during attacks. Cephalalgia 26(9):1071-79, 2006.
37. Chen F, Demers LM, Shi X. Upstream signal transduction of NF-kappaB activation. Curr Drug Targets Inflamm Allergy 1(2):137-49, 2002.
38. Tian B, Brasier AR. Identification of a nuclear factor kappa B-dependent gene network. Recent Prog Horm Res 58:95-130, 2003.
39. Connelly L, Palacios-Callender M, Ameixa C, Moncada S, Hobbs AJ. Biphasic regulation of NF-kappa B activity underlies the pro- and anti-inflammatory actions of nitric oxide. J Immunol 166(6):3873-81, 2001.
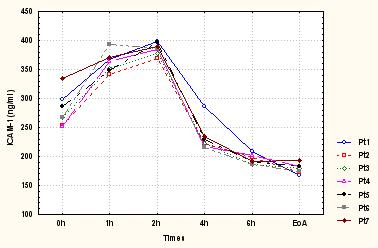
40. Sarchielli P, Alberti A, Baldi A, et al. Proinflammatory cytokines, adhesion molecules, and lymphocyte integrin expression in the internal jugular blood of migraine patients without aura assessed ictally. Headache 46(2):200-7, 2006.
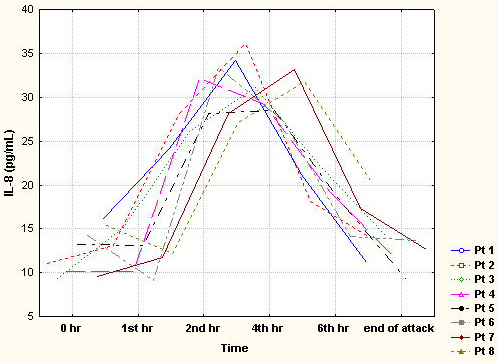
41. Sarchielli P, Alberti A, Vaianella L, et al. Chemokine levels in the jugular venous blood of migraine without aura patients during attacks. Headache 44(10):961-8, 2004.
42. Tran MT, Ritchie MH, Lausch RN, Oakes JE. Calcitonin gene-related peptide induces IL-8 synthesis in human corneal epithelial cells. J Immunol 164(8):4307-12, 2000.
43. Amann R, Peskar BA. Anti-inflammatory effects of aspirin and sodium salicylate. Eur J Pharmacol 447(1):1-9, 2002.
44. Vogler BK, Pittler MH, Ernst E. Feverfew as a preventive treatment for migraine: a systematic review. Cephalalgia 18:704-8, 1998.
45. Pfaffenrath V, Diener HC, Fischer M, Friede M, Henneicke-von Zepelin HH; Investigators. The efficacy and safety of Tanacetum parthenium (feverfew) in migraine prophylaxis--a double-blind, multicentre, randomized placebo-controlled dose-response study. Cephalalgia 22(7):523-32, 2002.
46. Diener HC, Pfaffenrath V, Schnitker J, Friede M, Henneicke-von Zepelin HH. Efficacy and safety of 6.25 mg t.i.d. feverfew CO2-extract (MIG-99) in migraine prevention--a randomized, double-blind, multicentre, placebo-controlled study. Cephalalgia 25(11):1031-41, 2005.
47. Bremner P, Heinrich M. Natural products as targeted modulators of the nuclear factor-kappaB pathway. J Pharm Pharmacol 54(4):453-72, 2002.
48. Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 8(2):136-42, 2002.
49. Dalkara T, Zervas NT, Moskowitz MA. From spreading depression to the trigeminovascular system. Neurol Sci 27 Suppl 2:S86-90, 2006.
50. Arulmani U, Maassenvandenbrink A, Villalon CM, Saxena PR. Calcitonin gene-related peptide and its role in migraine pathophysiology. Eur J Pharmacol 500(1-3):315-30, 2004.
51. Durham PL. Calcitonin gene-related peptide (CGRP) and migraine. Headache 46 Suppl 1:S3-8, 2006.
52. Durham P, Russo A. New insights into the molecular actions of serotonergic antimigraine drugs. Pharmacol Ther 94(1-2):77-92, 2002.
53. Peroutka SJ. Neurogenic inflammation and migraine: implications for the therapeutics. Mol Interv 5(5):304-11, 2005.
54. Cutrer FM, Moskowitz MA. Wolff Award 1996. The actions of valproate and neurosteroids in a model of trigeminal pain. Headache 36(10):579-85, 1996.
55. Akerman S, Goadsby PJ. Topiramate inhibits trigeminovascular activation: an intravital microscopy study. Br J Pharmacol 146(1):7-14, 2005.
56. Akerman S, Williamson DJ, Kaube H, Goadsby PJ. The effect of anti-migraine compounds on nitric oxide-induced dilation of dural meningeal vessels. Eur J Pharmacol 452(2):223-8, 2002.
57. Goldstein DJ, Offen WW, Klein EG, et al. Lanepitant, an NK-1 antagonist, in migraine prevention. Cephalalgia 21(2):102-6, 2001.
58. Diener HC; RPR100893 Study Group. RPR100893, a substance-P antagonist, is not effective in the treatment of migraine attacks. Cephalalgia 23(3):183-5, 2003.
59. May A, Gijsman HJ, Wallnofer A, Jones R, Diener HC, Ferrari MD. Endothelin antagonist bosentan blocks neurogenic inflammation, but is not effective in aborting migraine attacks. Pain 67(2-3):375-8, 1996.
60. Durham PL, Cady R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: implications for migraine therapy. Headache 44(1):35-42, 2004.
61. Edvinsson L. Calcitonin gene-related peptide (CGRP) and the pathophysiology of headache: therapeutic implications. CNS Drugs 15(10):745-53, 2001.
62. Taylor CK, Smith DD, Hulce M, Abel PW. Pharmacological characterization of novel CGRP receptor peptide antagonists that are selective for human CGRP receptors. J Pharmacol Exp Ther 2006 Jul 27 [Epub ahead of print]. DOI: 10.1124/jpet.106.108316.
63. Doods H, Hallermayer G, Wu D, et al. Pharmacological profile of BIBN4096BS, the first selective small molecule CGRP antagonist. Br J Pharmacol 129(3):420-3, 2000.
64. Hay DL, Poyner D. The preclinical pharmacology of BIBN4096BS, a CGRP antagonist. Cardiovasc Drug Rev 23(1):31-42, 2005.
65. Brain SD. Calcitonin gene-related peptide (CGRP) antagonists: blockers of neuronal transmission in migraine. Br J Pharmacol 142(7):1053-4, 2004.
66. Olesen J, Diener HC, Husstedt IW, et al; BIBN 4096 BS Clinical Proof of Concept Study Group. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med 350(11):1104-10, 2004.
67. Goadsby PJ. Calcitonin gene-related peptide antagonists as treatments of migraine and other primary headaches. Drugs 65(18):2557-67, 2005.
68. Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol 58(5):698-705, 2005.