Volumen 12, Número 1, Septiembre 2003
| |
Volumen 12, Número 1, Septiembre 2003 |
|
Expertos Invitados
El descubrimiento del receptor del fibrinógeno plaquetario (GPIIb/IIIa) fue promisorio acerca del desarrollo final de un agente antiagregante de las plaquetas.1 Dado que la unión del fibrinógeno es un requisito indispensable para la agregación plaquetaria, las drogas que la inhiben fueron consideradas el más poderoso y efectivo agente antiplaquetario que podía esperarse. Se utilizaron tres estrategias para desarrollar los antagonistas GPIIb/IIIa. La primera fue los anticuerpos monoclonales. El anticuerpo 7E3 fue elaborado en humanos y el fragmento fab se comercializó como abciximab.2 La segunda estrategia utilizó péptidos de veneno de víbora, que contienen alrededor de 70 aminoácidos (desintegrinas) y son muy potentes antagonistas de esta GP.3 Se incorporaron al comercio como eptifibatide, un pequeńo péptido cíclico.4 Si bien ambas estrategias producen muy potente efecto, no son activas por vía oral y solamente pueden ser administradas por vía endovenosa. Por lo mismo, su uso en forma rápida parece ser el único posible. Como se consideraba que el tratamiento ideal debía ser oral, varias empresas desarrollaron pequeńas moléculas inhibidoras. La primera fue el tirofiban.5 A pesar de que se trata de una pequeńa molécula no peptídica, no resultó ser activa a través de la vía deseada y actualmente se la emplea como compuesto intravenoso. Han sido elaborados diversos compuestos activos por vía oral, entre los que se cuentan xemilofiban,6 orbofiban,7 lotrafiban8 y sibrafiban.9 Los estudios clínicos favorecen el uso de inhibidores de la GPIIb/IIIa. El abciximab, eptifibatide y tirofiban mostraron beneficios en el tratamiento de síndromes agudos coronarios (SAC)10 y como adyuvantes en la angioplastia coronaria (AC).11 Hubo gran expectativa cuando estos compuestos de administración oral ingresaron en ensayos clínicos. A pesar de ser más potentes que sus contrapartidas endovenosas, estudios como EXCITE (xemilofiban),12 OPUS (orbofiban),13 BRAVO (lotrafiban)14 y SYMPHONY (sibrafiban)15,16 no mostraron beneficios en el tratamiento. De hecho, hubo tendencia hacia mayor número de eventos en los grupos de tratamiento.17 Esto plantea un problema muy desconcertante: żCómo puede una droga administrada por vía oral no ofrecer beneficios cuando se compara con otra similar dada por vía endovenosa? Objetivo Biodisponibilidad Dosis Agonismo parcial Recientemente ha habido una progresiva toma de conciencia sobre la
importancia de la GPIIb/IIIa mediada por seńales de adentro hacia
fuera. Es claro que los dominios citoplasmáticos de la glucoproteína
son importantes en la transducción de seńales.24 El
receptor ha demostrado también poseer actividad de isomerasa de
disulfuro.25 Esta capacidad para reubicar las uniones
disulfuro puede ser esencial en los procesos de seńalización. La unión
del fibrinógeno no es el paso final en la agregación, mientras que la
seńal generada por la unión de éste también es fundamental. La capacidad de seńalización del GPIIb/IIIa puede ser importante en
el fracaso de los agentes orales. La seńalización mediada por el
receptor sobre sí mismo parece tener escaso efecto, como los
antagonistas de la GPIIb/IIIa no tienen efectos detectables sobre la
función plaquetaria a cualquier dosis. No obstante, en la presencia de
un agonista débil tal como una concentración umbral de ADP, bajas
concentraciones de los inhibidores causan aumento en la agregación
plaquetaria.21 Farmacológicamente, esto ha sido descripto
como agonismo parcial. Sin embargo sería más adecuado describirlo como
un sistema de función dual de receptor con alto porcentaje de
receptores de repuesto. GPIIb/IIIa media la ligadura cruzada de
plaquetas vía fibrinógeno y también genera seńales de activación.
El fibrinógeno, como molécula divalente, tiene tanto la capacidad de
ligar en forma cruzada dos receptores como generar una seńal mientras
que los antagonistas pueden activar el receptor pero no realizar la unión
cruzada. Así, hay agonistas completos para la seńalización y
antagonistas completos para la agregación. Hay elevado porcentaje de
receptores de repuesto, y la ocupación de un número relativamente
pequeńo de receptores por el fibrinógeno es suficiente para provocar
la agregación plaquetaria. Igualmente, la ocupación de unos pocos
receptores es suficiente para disparar un máximo de seńales. Este
mecanismo es responsable de la estrecha relación dosis-respuesta con
estas drogas. Sitios de unión regulados por ligandos Es posible dividir a estos antagonistas en dos grupos, según sus efectos sobre los SURL. En un grupo está el abciximab, que no tiene actividad SUAL, en tanto que todos los inhibidores de moléculas pequeńas, tanto IV como orales, están en el otro grupo como mediadores de la actividad SUAL.31 Todas las drogas clínicamente utilizadas tienen baja actividad SUIL, sugiriendo que ésta puede no tener participación en los efectos protrombóticos de los antagonistas de la GPIIb/IIIa. Los datos clínicos confirman esta división, ya que el abciximab ha demostrado fehacientemente mayor eficacia que las moléculas pequeńas, aun los compuestos IV.32 Tal cosa indica que la inducción de SUAL puede ser la clave de los problemas con los antagonistas de la GPIIb/IIIa. Farmacocinética y antagonistas de la GPIIb/IIIa Qué anduvo mal con los antagonistas GPIIb/IIIa Futuro de los antagonistas de la GPIIb/IIIa orales
|
|
| |
Introducción Pacientes y métodos Se estudiaron 6 grupos de pacientes: Las muestras de sangre para la determinación de los niveles plasmáticos de ANF fueron obtenidas al inicio y a los 6, 18 y 24 meses. La extracción del plasma y el RIA se realizaron según el método descripto por Sarda y col.12 Los resultados se analizaron utilizando el test t de Student, con ajuste mediante recta de regresión y test de c2. Se consideró significativa una p < 0.05. Resultados Las concentraciones plasmáticas de ANF en los 6 grupos estudiados y a los distintos tiempos se observan en la tabla 1.  En las primeras 3 muestras (18 meses) no hubo diferencias significativas entre los pacientes chagásicos comparados con sus controles, o sea entre pacientes asintomáticos y los controles sanos; y entre los pacientes con TC y MCP chagásicos y no chagásicos. Como no se encontraron diferencias entre los pacientes infectados y los no infectados, se unieron los grupos y se los comparó según la gravedad de la patología: grupo I (controles sanos y chagásicos asintomáticos); grupo TC; y grupo MCP. La figura 1 muestra los resultados obtenidos comparando los niveles de ANF en los grupos a tiempos 0, 6, 18 y 24 meses. Los pacientes del grupo MCP presentaron niveles significativamente aumentados con respecto al grupo I y TC: figura 1a (t = 0): 67% y 64%, p < 0.001; figura 1b (t = 6): 53%, p < 0.001 y 43%, p<0.01; y 1c=66%, p<0.001 y 58%, p<0.01). A los 24 meses se observó aumento significativo en los valores de ANF plasmático en los pacientes asintomáticos (grupo I - Chagas) y con TC con respecto a los controles sanos (figura 1d, 46% y 71%, p < 0.01); y en los pacientes con MCP con respecto a los otros grupos (89%, p < 0.001 vs. control; 80%, p < 0.001 vs. grupo I - Chagas; y 62%, p < 0.01 vs. TC).  Figura
1. Factor natriurético atrial (ANF) plasmático. Grupo I
(controles sanos + pacientes asintomáticos con enfermedad de
Chagas). Grupo TC (pacientes con trastornos de la conducción
chagásicos y no chagásicos). Grupo MCP (pacientes con
miocardiopatía chagásicos y no chagásicos). 1a. Muestra
inicial (tiempo = 0). 1b. Muestra tiempo = 6 meses. 1c.
Muestra tiempo = 18 meses. (**) p < 0.001 vs.
grupo I. (*) p < 0.002 vs. grupo I, (x) p < 0.01 vs.
grupo TC. 1d. Muestra tiempo = 24 meses: grupo I -
control (controles sanos), grupo I - Chagas (pacientes asintomáticos),
(x) p < 0.01 vs. grupo I - control, (#) p < 0.01 vs.
grupo TC, (xx) p < 0.001 vs. grupo I - control y vs.
grupo I - Chagas. Figura
1. Factor natriurético atrial (ANF) plasmático. Grupo I
(controles sanos + pacientes asintomáticos con enfermedad de
Chagas). Grupo TC (pacientes con trastornos de la conducción
chagásicos y no chagásicos). Grupo MCP (pacientes con
miocardiopatía chagásicos y no chagásicos). 1a. Muestra
inicial (tiempo = 0). 1b. Muestra tiempo = 6 meses. 1c.
Muestra tiempo = 18 meses. (**) p < 0.001 vs.
grupo I. (*) p < 0.002 vs. grupo I, (x) p < 0.01 vs.
grupo TC. 1d. Muestra tiempo = 24 meses: grupo I -
control (controles sanos), grupo I - Chagas (pacientes asintomáticos),
(x) p < 0.01 vs. grupo I - control, (#) p < 0.01 vs.
grupo TC, (xx) p < 0.001 vs. grupo I - control y vs.
grupo I - Chagas.Para analizar la utilidad de los niveles del ANF como marcadores pronósticos en las MCP se estudiaron sus concentraciones en los pacientes que fallecieron durante el período de estudio (tabla 2).  Se correlacionaron los niveles de ANF en la primera muestra de sangre que se les extrajo (t = 0) con la causa de deceso (ICC o muerte súbita) producida durante los 24 meses de seguimiento. Fallecieron 16 pacientes con MCP (9 chagásicos); teniendo en cuenta el número de casos, se lo dividió en cuartilos: Q0.25 = 59.46 pg/ml; Q0.50 (mediana) = 84.96 pg/ml; y Q0.75 = 111.6 pg/ml. Se utilizó el estadístico c2 (confianza = 95%) para determinar si la distribución de muertes por encima y por debajo de cada uno de estos niveles era equitativa o si existían diferencias significativas. Los pacientes con MCP, chagásica o no chagásica, con niveles de ANF circulantes superiores a 111.6 pg/ml tuvieron una proporción de fallecimientos mayor (c2 = 4.40, p < 0.03) que los que presentaron niveles inferiores a esa cifra. Considerando la concentración de 111.6 pg/ml, el riesgo relativo (RR) fue 2.167, o sea que el grupo de pacientes que inicialmente tenían niveles iguales o superiores a esa cifra tuvo el doble de mortalidad que los que presentaron un nivel inferior. Discusión Existen muy pocos trabajos relacionados con los péptidos natriuréticos en la infección por T. cruzi y todos describen modelos experimentales de la enfermedad de Chagas. Piazza y col. 13 encontraron disminución en el contenido de ANF en extractos atriales durante la fase aguda de la infección; y Caliari y col.14 hallaron alteraciones morfológicas y morfométricas en los gránulos atriales, analizados por microscopía electrónica, al estudiar perros con insuficiencia cardíaca en el mismo período. Por otra parte en un trabajo previo10 demostramos la presencia de alteraciones morfológicas del miocardio y niveles aumentados de ANF en plasma tanto en el período agudo como en el crónico de la infección por T. cruzi en ratas. En este estudio se analiza por primera vez el nivel de ANF plasmático en pacientes con patologías cardíacas de origen chagásico. La infección por T. cruzi en los seres humanos presenta distintos estadios. Solamente el 3% de las personas infectadas desarrollan una miocarditis aguda después de infectarse, mientras que el 97% restante no muestra ningún síntoma de la enfermedad. Después de 10-20 ańos, el 30% de ellos desarrolla la MCP crónica chagásica. Hasta que aparecen las alteraciones cardiovasculares los pacientes permanecen en el período llamado asintomático o indeterminado, siendo muy difícil determinar el comportamiento del ANF en la etapa aguda de la infección. Por lo tanto, sólo se pudieron estudiar los que estaban en la fase crónica de la enfermedad. El ANF plasmático se encontró aumentado en los pacientes chagásicos, pero el incremento fue similar al hallado en pacientes con enfermedades cardíacas de otra etiología. Por lo tanto, los niveles aumentados del péptido no tendrían un origen inmunológico ni serían una consecuencia de la acción directa del parásito sobre la célula miocárdica; sino que derivarían de la función alterada del corazón, especialmente durante el estado dilatado. Por otra parte, cuando analizamos los niveles de ANF en pacientes chagásicos asintomáticos comparados con los controles sanos, encontramos que estos eran similares en las 3 primeras muestras pero estaban aumentados en la muestra obtenida a los 24 meses de comenzado en estudio; por lo tanto, el análisis de los niveles del péptido podría ser utilizado como factor pronóstico del desarrollo de miocardiopatía en pacientes asintomáticos en el largo plazo. Además, los pacientes chagásicos con niveles de ANF en plasma mayores de 111.6 pg/ml en la muestra inicial tuvieron el doble de mortalidad que los que presentaban valores inferiores del péptido. En conclusión, en este estudio se mostró la utilidad de los niveles plasmáticos de ANF como marcador de compromiso hemodinámico gradual en pacientes con MCP chagásica, ya evidenciado en pacientes con MCP de otros orígenes.14,15 Por otra parte, los niveles aumentados del péptido podrían ser utilizados como marcador precoz del desarrollo futuro de la MCP chagásica en los pacientes asintomáticos; mientras que las concentraciones iniciales iguales o superiores a 111.6 pg/ml del mismo serían un indicio de pronóstico adverso de sobrevida en los pacientes con MCP. BIBLIOGRAFIA
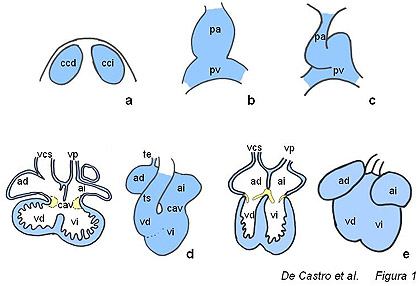
La formación del corazón es un
proceso complejo en el cual intervienen distintos tipos celulares
(figura 1). En las primeras fases de desarrollo embrionario, dos
regiones simétricas conspicuas del mesodermo se diferencian en
tejido promiocárdico, expresando así una serie de factores de
transcripción que invariablemente las convierten en
miocardiocitos. Estas dos regiones promiocárdicas, denominadas
crestas cardíacas, se fusionan en la línea media del embrión en
desarrollo y dan lugar a un tubo cardíaco inicial, compuesto de
dos capas; una capa interna de endocardio rodeada por una capa
externa de miocardio. Entre estas dos capas se localiza una
sustancia amorfa y acelular a modo de lámina basal denominada
gelatina cardíaca.10 Figura 1. Esquema ilustrativo de las diferentes etapas del desarrollo cardiaco. a, crestas precardíacas; b, tubo cardíaco inicial; c, asa cardíaca; d, corazón embrionario, en el cual se observa el inicio del proceso de septación de las cámaras atriales y ventriculares; e, corazón fetal, en el cual se observa que el proceso de septación se ha completado y las válvulas atrioventriculares se han desarrollado. ad, atrio derecho; ai, atrio izquiedo; cav, canal atrioventricular; ccd, cresta cardíaca derecha; cci, cresta cardíaca izquierda; pa, polo arterial; pv, polo venoso; te, tracto de entrada; td, tracto de salida; vcs, vena cava superior; vp, vena pulmonar; vd, ventrículo derecho; vi, ventrículo izquierdo. Según se avanza en el desarrollo, el tubo cardiaco inicial se delamina del mesodermo dorsal y sufre invariablemente una torsión hacia la derecha. Es a partir de este estadio cuando las futuras regiones ventriculares y atriales empiezan a vislumbrase a nivel morfológico, y como veremos más adelante también a nivel molecular. En este estadio embrionario se pueden distinguir cinco regiones claramente delimitadas: el tracto de entrada, las cámaras atriales, el canal atrioventricular, las cámaras ventriculares y el tracto de salida. Es también en este estadio cuando se empieza a observar los primeros signos de septación, dado que entre las cámaras atriales y ventriculares ya se empiezan a esbozar los futuros septos interatrial (septo interatrial primario) e interventricular (porción muscular), respectivamente. Curiosamente, existe una continuidad morfológica entre las regiones del tracto de entrada, canal atrioventricular y tracto de salida a nivel de la curvatura interna, lo cual ha llevado a plantear la hipótesis de que la especificación molecular del miocardio atrial y ventricular acontece exclusivamente en las regiones de la curvatura externa.3 Finalmente cabe resaltar que una de las etapas decisivas durante la morfogénesis cardíaca es la formación y alineamiento de los distintos septos cardíacos, para generar así un órgano pulsátil con doble circuito sanguíneo. De este modo, el corazón embrionario sufre una tabicación del tracto de entrada, canal atrioventricular y tracto de salida que permite obtener entradas y salidas independientes a las cámaras atriales y ventriculares derecha e izquierda, respectivamente. En este estadio se configura también a nivel morfológico el sistema de conducción cardíaco, el cual permite que se produzca la contracción sincrónica y acompasada de las distintas cámaras cardíacas.9 En los últimos ańos hemos experimentado un gran avance en el conocimiento de los mecanismos moleculares que regulan la cardiogénesis. Hemos asistido al descubrimiento de un número importante de factores de transcripción específicos del miocardio, así como se ha establecido la amplia heterogeneidad en proteínas estructurales que subyace al miocardio en formación y que se mantiene en la mayoría de los casos en el corazón adulto.5,6,8 En esta revisión, queremos realizar un barrido por los distintos patrones de expresión que se observan en el miocardio en desarrollo, así como en el adulto, e ilustrar cuáles son las implicaciones que dicha heterogeneidad provoca en la práctica clínica. Para ello es importante resaltar cuáles son los mecanismos de especificación celular que acontecen durante el desarrollo. Las células de mesodermo en formación reciben seńales de los tejidos contiguos en forma de factores de crecimiento secretados que, mediante la activación de receptores de membrana específicos, provocan directa o indirectamente que unos factores de transcripción se transcriban y por tanto sean activos. Estos factores de transcripción generan a su vez respuestas en el núcleo que conllevan a la síntesis de proteínas estructurales, tales como proteínas contráctiles, del citoesqueleto o reguladoras del impulso eléctrico. Así pues, la generación de distintos patrones de expresión de los factores de transcripción, proteínas contráctiles/citoesqueleto y proteínas reguladores del impulso eléctrico es indicativa, respectivamente, de características morfogéneticas, estructurales y funcionales del corazón en formación. En los primeros estadios del desarrollo cardíaco, distintos factores de transcripción tales como MEF2C, Nkx2.5 y GATA4, entre otros, muestran un patrón de expresión homogéneo a lo largo del tubo cardíaco inicial.8 Otros factores de transcripción muestran sin embargo una regionalización en su expresión en los distintos ejes embrionarios; Tbx5 muestran un gradiente ántero- posterior,3 Hand2 (eHAND) muestra una expresión diferencial en el eje dorso- ventral1 y Pitx2 se expresa exclusivamente en la región izquierda del eje derecha- izquierda.2 Dentro de las proteínas estructurales, distintas proteínas sarcoméricas muestran diferencias en el eje ántero-posterior pero no hay evidencias de regionalización en el eje dorso-ventral. En el caso del eje derecha-izquierda, existe una proteína de la matriz extracelular, la flectina, que se expresa exclusivamente en la región izquierda.14 Por el contrario, no existen evidencias de expresión de ninguna de las proteínas involucradas en la generación o conducción del impulso eléctrico en este estadio (figura 2A).  
Figura 2. Esquemas representativos de los diferentes patrones de expresión en las distintas etapas del desarrollo cardiaco: A, etapa de tubo cardíaco inicial; B, etapa embrionaria; y C, etapa fetal/adulto. En la etapa de tubo cardíaco inicial (A) existe una regionalización en los tres ejes embrionarios. En el eje anteroposterior (A/P) muestran heterogeneidad en su expresión factores de transcripción tales como Tbx5 y proteínas sarcoméricas tales como αMHC y βMHC, así como proteínas involucradas en el metabolismo del calcio, tales como SERCA2 a y fosfolamban (PBL). En el eje derecha/izquierda (L/R) muestran una expresión diferencial el factor de transcripción Pitx2 y la proteína extracelular flectina. En el eje dorso- ventral (D/V) sólo el factor de transcripción Hand2 muestra diferencias de expresión. En la etapa embrionaria (B) existe una mayor heterogeneidad en la expresión. Hay genes que muestran expresión sólo en las regiones de formación de miocardio (i; p.ej. GATA5 y GATA6), genes que muestran diferencias entre el miocardio primario y el miocardio de cámara (ii; SERCA2a, PLB, Tbx2, BMP2, BMP4, por mencionar algunos), genes que se expresan tan sólo en las cámaras atriales o en las ventriculares (iii; p. ej. αMHC, βMHC, MLC2a y MLC2v), genes que muestran expresión en el miocardio derivado de la cresta cardíaca izquieda (iv; Pitx2) o genes que parecen demarcar el futuro sistema de conducción ventricular (v; Irx2, Irx3 y Tbx3). Finalmente, en la etapa adulta, la heterogeneidad molecular parece restringirse al miocardio auricular puesto que el miocardio ventricular muestra sólo expresión en gradiente a lo largo de las paredes libres ventriculares. En el miocardio auricular existe heterogeneidad molecular entre el miocardio de las venas cavas (i), el miocardio mediastinal (ii), el miocardio derivado del canal atrioventricular embrionario (iii) y el miocardio de las aurículas propiamente dicho (iv). En el estadio embrionario, la mayoría de los factores de
transcripción muestran un patrón homogéneo, tales como los
anteriormente mencionados y otros, tales como GATA5 y GATA6, solo
se expresan en aquellas zonas donde se está produciendo aún el
reclutamiento de nuevas células miocárdicas.12 En
este estadio existe un refinamiento de la diferenciación
dorso-ventral de tal modo que algunos factores de transcripción
delimitan las fronteras entre el miocardio primario y el miocardio
específico de cámara (atrial y ventricular), como es el caso de
Tbx2.10 A su vez aparecen las primeras diferencias de
expresión entre el miocardio sistémico y pulmonar, ilustrados
por Hand1 y Hand2.13 También aparecen los primeros
signos de diferenciación del sistema ventricular de conducción,
con la expresión de distintos factores de transcripción, Irx2,
Irx3 y Tbx3, en las presuntas áreas de formación del fascículo
de His y las ramas derecha e izquierda.4 Las
diferencias en el eje derecha-izquierda siguen estando
representadas por Pitx2, resaltado que dichas diferencias se
mantiene como tales en las cámaras atriales pero son convertidas
en diferencias dorso-ventral en las cámaras ventriculares.2
Los patrones de expresión de las proteínas contráctiles
refuerza la heterogeneidad molecular en el eje ántero-posterior y
en las regiones de miocardio primario/cámara (miosinas y
actinas), así como en los recién creados compartimientos sistémico/pulmonar
(miosinas).16 Dicha heterogeneidad en la expresión de
proteínas contráctiles también se extiende al sistema
ventricular de conducción cardiaca.
|
Trabajos Distinguidos, Serie Cardiología,
integra el Programa SIIC de Educación Médica Continuada
![]()
[Bienvenidos
a siicsalud]
[Acerca de SIIC] [Estructura de SIIC] [Círculo SIIC de Lectores]
[Salud(i)Ciencia]
[Trabajos Distinguidos]
Sociedad
Iberoamericana de Información Científica (SIIC)
Av. Belgrano 430, (C1092AAR), Buenos Aires, Argentina
Correo electrónico (e-mail): atencionallector@siicsalud.com;
Tels: +54 11 4342-4901; Fax: +54 11 4331-3305.
Correo SIIC: Casilla de Correo 2568, (C1000WAZ) Correo Central, Buenos Aires.
Copyright siicsalud©
1997-2003, Sociedad Iberoamericana de Información Científica
(SIIC)