Introducción
La enfermedad coronaria es una de las causas principales de enfermedad y muerte. La mayoría de los síndromes coronarios agudos son consecuencia de la formación de trombos intravasculares sobreimpuestos a la rotura o erosión de una placa en las arterias c
oronarias. La glucoproteína plaquetaria IIb/IIIa es un receptor de membrana con un papel principal en la adhesión plaquetaria y, básicamente, en la formación del trombo.1 Desde el primer informe del alelo PIA2 como factor de riesgo
para la enfermedad coronaria,2 estudios numerosos y contradictorios evaluaron la asociación entre los polimorfismos de esta glucoproteína y la trombosis arterial. En esta revisión, seńalamos lo que conocemos acerca de la hipótesis de que los p
olimorfismos en diferentes glucoproteínas plaquetarias (GP) (IIIa, Ia, Ibα y VI) incrementan el riesgo de infarto miocárdico o enfermedad cardiovascular isquémica.
Polimorfismo de la glucoproteína plaquetaria GP IIb/IIIa PIA1/PIA2
La glucoproteína plaquetaria IIb/IIIa es un miembro de la familia de las integrinas, moléculas adhesivas que mediante la unión con el fibrinógeno y el factor de Von Willebrand, promueven la agregación y la trombosis.3,4 El complejo G
P IIb/IIIa constituye el receptor del fibrinógeno, cuyo engranaje representa la vía final común para la agregación plaquetaria.5 Cuantitativamente es la más abundante y funcionalmente es la más importante de las integrinas en las plaquetas. El
polimorfismo PIA1/PIA2, también llamado HPA-1a/HPA-1b, resulta de una sustitución de leucina (alotipoA1) en prolina (alotipoA2) en la posición 33 de la subunidad β3 de la GP IIb/IIIa.6 El alelo
más común PIA1 se encuentra en aproximadamente el 75% al 80% de la población de raza blanca y el alelo PIA2 en el 15% al 20%, con cerca de un 2% de portadores homocigotas PIA2/A2.7,8
Impacto del polimorfismo PlA1/PlA2 sobre el proceso trombótico in vitro
Estudios sobre los mecanismos por los cuales el PIA2 podría contribuir a la trombosis arterial informaron resultados contradictorios: en 1999, Feng y colaboradores demostraron un pequeńo pero significativo aumento de la agregabilidad plaqu
etaria asociada con el alelo PIA2 de una forma dependiente de la dosis génica. En ese estudio, la agregabilidad plaquetaria fue evaluada in vitro mediante agregometría e inducida por epinefrina.9 El alotipo PIA2 se
asoció con una mayor adhesión con el fibrinógeno, con un umbral más bajo para la activación plaquetaria,10 con una mayor retracción del coágulo11 y con una mejoría en la formación de trombina.12 Andreoli y colaboradores
no encontraron diferencias entre la agregación plaquetaria de PIA1 y PIA2 cuando se utilizó trombina o ADP como estimuladores.13 Sin embargo, la agregación plaquetaria PIA2 fue reducida en comparación con las p
laquetas PIA1 al utilizar ácido araquidónico como agonista. Para este autor, el alelo PIA2 parece asociado con una deficiencia funcional en las plaquetas, específicamente ligada a la activación del receptor del fibrinógeno por medio
del tromboxano A2.
Sin embargo, para Cadroy y colaboradores, el polimorfismo PIA1/PIA2 no influyó la formación de trombos en estudios experimentales arteriales y el efecto potencial de este polimorfismo sobre la trombogénesis arterial depende de las c
ondiciones del experimento, el flujo sanguíneo local y la tracción de cizallamiento.14,15 Debido a que los estudios realizados in vitro no reflejan necesariamente la amplia gama de la respuesta funcional plaquetaria observada in vivo
, la influencia funcional del polimorfismo PIA1/PIA2 sobre la formación del trombo no está claramente demostrada hoy en día.
Impacto clínico del polimorfismo PIA1/PIA2
En 1996, Weiss y colaboradores demostraron un aumento en el riesgo de infarto de miocardio (IM) o angina inestable en portadores del alelo PIA2 del gen de la glucoproteína IIIa, en una cohorte de 71 pacientes.2 El odds
ratio (OR) para el desarrollo de un evento coronario fue de 2.8 con un intervalo de confianza de 95% (IC 95%) comprendido entre 1.2 y 6.4, entre sujetos con polimorfismo PIA2. Esta asociación fue más fuerte entre aquellos pacientes con eve
ntos coronarios antes de los 60 ańos (OR 6.6; IC 95% 1.8 a 22.4). Carter y colaboradores encontraron también una asociación significativa entre el polimorfismo PIA2 y el IAM con un vínculo particularmente fuerte en hombres menores de 47 ańos e
n quienes se halló una incidencia de 50% del alelo PIA2 comparado con un 27% en sujetos control equiparados en cuanto a edad y sexo.16 La expresión clínica de este polimorfismo parece también estar reforzada por el tabaquismo.1
7 Sin embargo, en el mismo ańo, un estudio prospectivo realizado en una cohorte de 14 916 pacientes demostró un resultado opuesto sin una relación significativa entre el alelo PIA2 y el riesgo de IAM (OR 0.93; IC 95% 0.7 a 1.2).18
Para resolver esta controversia se realizaron numerosos estudios y metaanálisis con estratificación sobre factores de riesgo tradicionales, género y edad (Tablas 1 y 2).

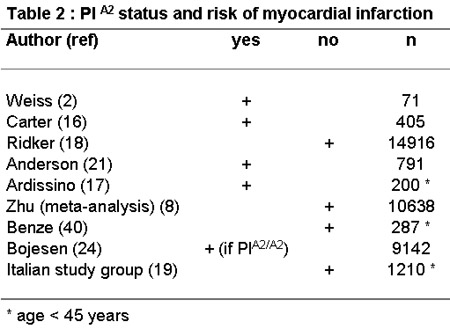
En un metaanálisis publicado en 2000, el polimorfismo PIA2 no estuvo asociado con un riesgo aumentado para el desarrollo de IA (n = 10 638; OR global 1.06, IC 95% 0.97 a 1.16).8 En un estudio reciente y grande, para los pacientes qu
e sobrevivieron a un primer IAM a una edad menor de 45 ańos el alelo PIA2 no fue estadísticamente más frecuente que en los controles emparejados (OR 0.9; IC 95%: 0.8 a 1.2).19 Luego de un primer episodio de IAM el genotipo PIA1
/A2 confirió un riesgo relativo de 1.38 (IC 95%: 1.04 a 1.83) para un evento recurrente (muerte debida a enfermedad coronaria o IAM no fatal) en el lustro siguiente.20
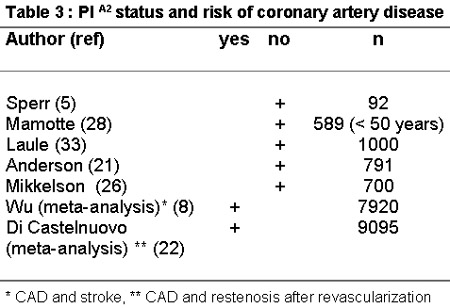
Al considerar la asociación del polimorfismo PIA2 con la enfermedad vascular en forma global (coronariopatía con IAM o sin IAM, eventos adversos luego de la angioplastia coronaria o accidente cerebrovascular) en la población general, la asocia
ción es significativa pero débil con el OR tomado en conjunto entre 1.10 y 1.12.21-23 (Tabla 3).
Cuando el criterio de valoración primario compuesto IAM o accidente cerebrovascular se considera no hay aumento del riesgo entre los heterocigotas PIA1/A2 contra los homocigotas PIA1/A1. Sin embargo, en Dinamarca, en una investigaci
ón prospectiva en la población general la homozigosidad PIA2/A2 contra PIA1/A1 se asoció con un riesgo tres veces mayor de enfermedad isquémica.24 Este riesgo no se observa en mujeres ni con el incremento de la edad en lo
s hombres jóvenes: respectivamente, los riesgos relativos ajustados en forma multifactorial en hombres menores de 40 ańos, de 40 a 50 ańos y en mayores de 50 ańos fueron 3.0 (1.1 a 8.0), 2.0 (1.0 a 3.9) y 1.0 (0.6 a 1.8). Otro estudio sugiere que el alel
o PIA2 es más frecuente en infartos cerebrales asociados con la oclusión de grandes vasos,1 pero no hay asociación entre el polimorfismo PIA1/A2 y el grosor de la capa íntima-media de la carótida.25
La limitación principal de estos estudios es que sólo se incluyeron pacientes que sobrevivieron a IAM. En una serie prospectiva de autopsias, Mikkelsson y colaboradores estudiaron 700 hombres de mediana edad (de 33 a 70 ańos) que padecieron muerte súbita
extrahospitalaria. Estos estudios sugieren que los homocigotas PIA1/A1 pueden ser más propensos a aterosclerosis temprana y a una progresión más rápida de enfermedad coronaria estable mientras que los portadores del alelo PIA2 son
más propensos a la trombosis aguda fatal.26, 27
El polimorfismo PIA2 y el riesgo de eventos adversos luego de un procedimiento de intervención coronaria mediante cateterismo
También existen algunas controversias sobre el papel del polimorfismo PIA1/A2 en la presencia de eventos adversos luego de intervención de las arterias coronarias mediante cateterismo28-33 (Tabla 4).

El alelo PIA2 no está asociado con riesgo significativamente elevado de eventos coronarios luego de la angioplastia coronaria con balón28 pero otros estudios documentaron una asociación entre este polimorfismo y trombosis subaguda d
el stent 29 o desarrollo de reestenosis luego del implante del stent.30 Luego del implante intracoronario del stent, en una indicación de rescate, la incidencia de IA dentro de los 30 días siguientes es más alta
en pacientes con el alelo PIA2 (riesgo relativo de oclusión del vaso del stent 4.2%, IC 95%: 1.5 a 12).31 Este riesgo más alto de trombosis del stent quedó también demostrado por Kastrati y colaboradores en pacientes
homocigotas para el alelo PIA2 pero no para los heterocigotas PIA1/A2.32 La tasa de eventos acumulativos (IAM, muerte o revascularización del vaso blanco) luego de 6 meses no reveló diferencias entre pacientes con el alel
o PIA2 o sin él. También existe una relación entre el tratamiento con estatinas y el desarrollo de reestenosis con respecto al status del alelo PIA2. En la angiografía de seguimiento, las tasas de reestenosis se encontraron r
educidas en forma significativa por el tratamiento con estatinas en portadores del alelo PIA2 pero no para los pacientes homocigotas para PIA1.31
Interacciones entre el polimorfismo PIA1/PIA2 y los inhibidores plaquetarios
Diversos estudios demostraron in vitro mayor sensibilidad al ácido acetilsalicílico en plaquetas PIA1/A2 que en las plaquetas PIA1/A1.10,13,34 Recientemente determinamos en un ensayo clínico que el genotipo PI<
sup>A1/A1 es uno de los numerosos mecanismos involucrados en la resistencia a la aspirina.35 Las plaquetas PIA1/A2 mostraron también mayor sensibilidad para el abciximab10 y pueden modificar la respuesta para el orb
ofiban, un antagonista oral de la GP IIb/IIIa.36
A pesar de la abundante literatura, el impacto del polimorfismo PIA1/A2 sobre la formación de trombos y la presencia de enfermedad isquémica cardiovascular todavía es incierto. Las interacciones entre las drogas antiplaquetarias o las estatina
s pueden representar algunas de las discrepancias entre los estudios acerca del PIA1/A2 como factor de riesgo para el infarto de miocardio y para las complicaciones de los procedimientos intervencionistas a nivel coronario. Probablemente la pr
esencia del alelo PIA2 es un factor de riesgo independiente débil y más obvio en la minoría de los hombres jóvenes homocigotas PIA2/A2.
El polimorfismo de la glucoproteína plaquetaria Ia-IIa
La GP Ia-IIa es uno de los receptores principales del colágeno en las plaquetas y una mutación en el nucléotido 807 del gen α2 se asocia fuertemente con la variación de la expresión de la integrina α2β1 sobre la superficie plaquetari
a.37 En la literatura, el polimorfismo de la GP Ia C807T no es un factor de riesgo para la enfermedad coronaria.38 En una cohorte de 2 237 pacientes de sexo masculino a quienes se les realizó coronariografía, Santoso y col. no halla
ron relación entre la presencia del alelo C y el riesgo de infarto de miocardio. La asociación fue estadísticamente significativa sólo entre pacientes menores de 62 ańos, con el OR más alto entre los pacientes menores de 45 ańos.39 Sin embargo
, esta asociación entre el alelo T807 con IAM entre los individuos más jóvenes se confirma en dos estudios recientes.19,40 Moshfegh y col.41 hallaron que el genotipo homocigota 807TT confirió un aumento de 3.3 veces en el riesgo de
IAM, pero Croft y col. no lo confirmaron en pacientes homocigotas en una cohorte más grande.38 Las diferencias en la frecuencia del genotipo 807TT en los grupos de control pueden explicar estos resultados contradictorios. Anvari y colaboradore
s compararon las frecuencias del alelo 807T en 94 sobrevivientes de muerte súbita cardíaca con un grupo igualado de 106 pacientes con enfermedad coronaria sin muerte súbita y 217 individuos sanos. No se encontró una sobrerrepresentación del alelo en ning
ún grupo.42 La portación del alelo de la GP Ia 807T tampoco está asociada con riesgo aumentado de reestenosis o de resultados tardíos desfavorables luego del implante del stent coronario.43
El polimorfismo de la glucoproteína plaquetaria GP Ibα
El complejo del receptor plaquetario GP Ib-IX-V, que comprende cuatro polipéptidos, juega un papel principal en la adhesión plaquetaria mediante la unión con el factor de Von Willebrand (VWF). Se evaluaron tres polimorfismos en el foco de la enfermedad a
rterial coronaria (EAC):
El polimorfismo de la GP Ibα T/M con una sustitución Thr/Met en la posición 145.
El polimorfismo de la GP Ibα VNTR o el número variado de marcadores con repeticiones en tándem (cuatro variantes designadas de la A a la D, que determinan la distancia entre el VWF y la superficie de las plaquetas).
La GP Ibα 5T/c o secuencia de Kozac, que resulta de la posición de una timina o citosina en la posición -5 relativa al codón de inicio ATG y afecta la expresión del complejo GPIb/IX/V sobre la superficie plaquetaria.
Impacto del polimorfismo de la GP Ibα T/M: en un estudio grande de casos de autopsia en la población blanca finlandesa, los hombres con IAM o trombosis coronaria fueron más probablemente portadores del genotipo de la GP Ibα T/M que los
sujetos de control que fallecieron de causas no cardíacas (OR: 2.0 y 2.6 respectivamente, p< 0.005 para ambos).44
Impacto del polimorfismo de la GP Ibα VNTR: en una cohorte de pacientes japoneses, el alelo A mostró asociación con la enfermedad coronaria.45 En una población indoasiática y caucásica no se encontraron asociaciones entre el polim
orfismo de la GP Ibα VNTR y la presencia de IAM.46 Dos estudios demostraron una fuerte asociación entre los polimorfismos de la GP Ibα T/M y el VNTR (isoforma B) y sugirieron que este haplotipo puede considerarse como factor de rie
sgo mayor de trombosis coronaria, infarto miocárdico fatal y muerte súbita cardíaca en hombres de mediana edad.39,47
Impacto del polimorfismo de la GP Ibα Kozak: para determinar el papel del polimorfismo de la secuencia Kozak de GP Ibα como factor de riesgo potencial para la enfermedad de las arterias coronarias, Meisel y col. genotipificaron 1 000 p
acientes con coronariopatía documentada por angiografía, así como en 1 000 sujetos controles igualados en edad y género. Las frecuencias del alelo Kozak fueron de 18.2% en pacientes y 13.8% en los controles, con frecuencias homocigotas T/T de aproximadam
ente 2% en ambos grupos.48 La conclusión principal de este estudio fue que el alotipo Kozac no fue un factor de riesgo para enfermedad coronaria, pero que los portadores de este alelo tienen un mayor riesgo para el desarrollo de síndromes coro
narios agudos (OR 1.43, IC 95%: 1.05 a 1.95) y complicaciones luego de la angioplastia coronaria sin colocación de stent (OR 3.75, IC 95%: 1.15 a 12.27).
Esto se contradice con los resultados de otros estudios que demostraron que el genotipo Kozac T/C no fue un factor de riesgo para IAM y paradójicamente puede conferir protección relativa contra el IAM.49,50 En forma experimental, el papel del
polimorfismo de la secuencia Kozac sobre la formación de trombos parece muy dependiente de la tasa del flujo sanguíneo.14
Polimorfismo de la glucoproteína plaquetaria GP VI
La glucoproteína VI es uno de los receptores plaquetarios principales para el colágeno y de importancia promordial en la activación y agregación plaquetarias mediante la activación de la fosfolipasa Cγ2. Croft y col. identificaron diez dimorfis
mos de la GP VI en un grupo de sujetos sanos y demostraron que el genotipo de la GP VI 13254CC aumenta el riesgo de IAM entre no fumadores, mujeres y personas mayores de 60 ańos en una cohorte de 525 pacientes con IAM y 474 controles, todos menores de 75
ańos. La heterocigocidad para el alelo de la GP VI 13254CC no se asoció con aumento en el riesgo de IAM en ninguno de los subgrupos estudiados.51
Conclusión
A pesar de la abundante literatura disponible, la significación clínica de los polimorfismos en las glucoproteínas receptoras en las plaquetas como factores de riesgo potenciales para la cardiopatía isquémica todavía no está clara. Los resultados fue
ron contradictorios o tuvieron significación estadística en el límite, debido a que los determinantes de la cardiopatía isquémica son multifactoriales y poligénicos. Estudios de investigación futuros determinarán cuáles polimorfismos son de importancia f
uncional o farmacológica y si esos polimorfismos son sólo marcadores de la enfermedad coronaria o factores de riesgo verdaderos.
BIBLIOGRAFÍA
-
Szolnoki Z, Somogyvari F, Kondacs A, Szabo M, Bene J, Havasi V, Komlosi K, Melegh B. Increased prevalence of platelet glycoprotein IIb/IIIa PlA2 allele in ischaemic stroke associated with large vessel pathology. Thromb Res 2003 ;109
:265-9.
-
Weiss EJ, Bray PF, Tayback M, Schulman SP, Kikler TS, Becker LC, Weiss JL, Gerstenblith G, Goldschmidt-Clermont PJ. A polymorphism of a platelet glycoprotein receptor as an inherited risk factor for coronary thrombosis. N Engl J Med 199
6; 334: 1090-4.
-
Phillips D, Charo I, Parise L, Fitzgerald L. The platelet membrane glycoprotein IIb/IIIa complex. Blood 1998;71:831-43.
-
Bray PF. Integrin and polymorphisms as risk factors for thrombosis. Thromb Haemost 1999 ;82 :337-44.
-
Sperr W, Huber K, Roden M, Janisiw M, Lang T, Graf S, Maurer G, Mayr W, Panzer S. Inherited platelet glycoprotein polymorphisms and risk for coronary heart disease in young central Europeans. Thromb Res 1998 ;90 :117-23.
-
Newman PJ, Derbes RS, Aster RH. The human platelets alloantigens, PlA1 and PlA2, are associated with a leucine33/proline33 amino acid polymorphism in membrane glycoprotein IIIa, and are distinguishable by DNA typing. J Clin Invest 1989 ;83 :17
78-81.
-
Undas A, Sanak M, Musial J, Szceklik A. Platelet glycoprotein IIIa polymorphism, aspirin, and thrombin generation. Lancet 1999 ;353 :982-3.
-
Zhu MM, Weedon J, Clark LT. Meta-analysis of the association of platelet glycoprotein IIIa PlA1/A2 polymorphism with myocardial infarction. Am J Cardiol 2000 ; 86 : 1000-5.
-
Feng D, Lindpaintner K, Larson M, Rao V, O'Donnell C, Lipinska I, Schmitz C, Sutherland P, Silbershatz H, D'Agostino R, Muller J, Myers R, Levy D, Tofler G. Increased platelet aggregability associated with platelet GPIIIa Pl A2 polymorphism. The Fram
ingham offspring study. Arterioscler Thromb Vasc Biol 1999 ;19 :1142-7.
-
Michelson A, Furman M, Goldschmidt-Clermont P, Mascelli MA, Hendrix C, Coleman L, Hamlington J, Barnard M, Kickler T, Christie D, Kundu S, Bray P. Platelet GPIIIa PlA polymorphisms dysplay different sensitivities to agonists. Circulatio
n 2000 ;101 :1013-8.
-
Vinod Vijayan K, Goldschmidt-Clermont PJ, Roos C, Bray PF. The PlA2 polymorphism of integrin ?3 enhances outside-in signalling and adhesive functions. J Clin Invest 2000 ;105 :793-802.
-
Andrioli G, Minuz P, Solero P, Pincelli S, Ortolani R, Lussignoli S, Bellavite P. Defective platelet response to arachidonic acid and thromboxane A2 in subjects with PlA2 polymorphism of b3 subunit (glycoprotein IIIa). Br J H
aematol 2000 ;110 :911-8.
-
Undas A, Brummel K, Musial J, Mann K, Szczeklik A. PlA2 polymorphism of ?3 integrins is associated with enhanced thrombin generation and impaired antithrombotic action of aspirin at the site of microvascular injury. Circulation 2001;104:26
66-72.
-
Cadroy Y, Sakariassan K, Charlet JP, Thalamas C, Boneu B, Sie P. Role of 4 platelet membrane glycoprotein polymorphisms on experimental arterial thrombus formation in men. Blood 2001 ;98 :3159-61.
-
Cadroy Y, Sakariassen K, Grandjean H, Thalamas C, Boneu B, Sie P. The effect of platelet PlA polymorphism on experimental thrombus formation in man depends on blood flow and thrombogenic substrate. Thromb Haemost 2001 ;85 :1097-103.
-
Carter A, Ossei-Geming N, Wilson I, Grant P. Association of the platelet PlA polymorphism of glycoprotein GPIIb/IIIa and the fibrinogen B? 448 polymorphism with myocardial infarction and extent of coronary artery disease. Circulation
1997 ;96 :1424-31.
Ardissino D, Manucci P, Merlini P, Duca F, Fetiveau R, Tagliabue L, Tubaro M, Galvani M, Ottani F, Ferrario M, Corral J, Margaglione M. Prothrombotic genetic risk factors in young survivors of myocardial infarction. Blood 1999 ;94 :46-51.
Ridker PM, Hennekens CH, Schmitz C, Stampfer MJ, Lindpaintner K. PlA1A2 polymorphism of platelet glycoprotein IIIa and risks of myocardial infarction, stroke and venous thrombosis. Lancet 1997 ;349 :385-8.
Atherosclerosis, Thrombosis, and Vascular Biology Italian Study Group. No evidence of association between prothrombotic gene polymorphisms and the development of acute myocardial infarction at a young age. Circulation 2003 ;107 :1117-22.
Bray P, Cannon C, Goldschmidt-Clermont P, Moyé L, Pfeffer M, Sacks F, Braunwald E. The platelet PlA2 and angiotensin-converting enzyme (ACE) D allele polymorphisms and the risk of recurrent events after acute myocardial infarction. Am J
Cardiol 2001 ;88 :347-52.
Anderson JL, King GJ, Bair TL, Elmer SP, Muhlestein JB, Habashi J, Carlquist JF. Association between a polymorphism in the gene encoding glycoprotein IIIa and myocardial infarction or coronary artery disease. J Am Coll Cardiol 1999 ;33 :727-33
.
Di Castelnuovo A, de Gaetano G, Donati MB, Iacoviello L. Platelet glycoprotein receptor IIIa polymorphism PlA1/A2 and coronary risk : a meta-analysis. Thromb Haemost 2001 ;85 :626-33.
Wu A, Tsongalis G. Correlation of polymorphisms to coagulation and biochemical risk factors for cardiovascular diseases. Am J Cardiol 2001 ;87 :1361-6.
Bojesen S, Juul K, Schnohr P, Tybjaerg-Hansen A, Nordestgaard B. Platelet glycoprotein IIb/IIIa PlA1/A2 homozygosity associated with risk of ischemic cardiovascular disease and myocardial infarction in young men. The Copenhagen city heart
study. J Am Coll Cardiol 2003 ;42 :661-7.
Garg U, Arnett D, Folsom A, Province M, Williams R, Eckfeldt J. Lack of association between platelet glycoprotein IIb/IIIa receptor PlA polymorphism and coronary artery disease or carotid intima-media thickness. Thromb Res 1998 ;89
:85-9.
Mikkelsson J, Perola M, Laippala P, Pentilla , Karhunen PJ. GPIIIa PlA1/A2 polymorphism and sudden cardiac death. J Am Coll Cardiol 2000 ;36 :1317-23.
Mikkelsson J, Perola M, Pentilla A, Goldsmidt-Clermont PJ, Karhunen PJ. The GPIIIa (beta(3) integrin) PlA polymorphism in the early development of coronary atherosclerosis. Atherosclerosis 2001 ;154 :721-7.
Mamotte C, van Bockxmeer F, Taylor R. PlA1/A2 polymorphism of glycoprotein IIIa and risk of coronary artery disease and restenosis following coronary angioplasty. Am J Cardiol 1998 ;82 :13-6.
Walter DH, Schächinger V, Elsner M , Dimmeler S, Zeiher AM. Platelet glycoprotein IIIa polymorphism and risk of coronary stent thrombosis. Lancet 1997 ;350 :1217-9.
Abbate R, Marcucci R, Camacho-Vanegas O, Pepe G, Gori AM, Capanni M, Simonetti I, Prisco D, Gensini GF. Role of platelet glycoprotein PlA1A2 polymorphism in restenosis after percutaneous transluminal coronary angioplasty. Am J Cardiol
. 1998;82 :524-5.
Walter D, Schachinger V, Elsner M, Mach S, Dimmeler S, Auch-Schwelk W, Zeiher AM. Statin therapy is associated with reduced restenosis rate after coronary stent implantation in carriers of the PlA2 allele of the platelet glycoprotein IIIa
gene. Eur Heart J 2001 ;22 :587-95.
Kastrati A, Koch W, Gawaz M, Mehilli J, Bottinger C, Schomig K, von Beckerath N, Schomig A. Pl polymorphism of glycoprotein IIIa and risk of adverse events after coronary stent placement. J Am Coll Cardiol 2000 ;36 :84-9.
Laule M, Cascorbi I, Stangl V, Bielecke C, Wernecke KD, Mrozikiewicz PM, Felix SB, Roots I, Baumann G, Stangl K. A1/A2 polymorphism of glycoprotein IIIa and association with excess procedural risk for coronary catheter interventions : a case-control
study. Lancet 1999 ;353 :708-12.
Cooke G, Bray P, Hamlington J, Pham D, Goldschmidt-Clermont P. PlA2 polymorphism and efficacy of aspirin. Lancet 1988 ;351 :1253-4.
Macchi L, Christiaens L, Brabant S, Sorel N, Ragot S, Allal J, Mauco G, Brizard A. Resistance in vitro to low-dose aspirin is associated with platelet PlA1 (GP IIIa) polymorphism but not with C807T (GP Ia/IIa) and C-5T Kozak (GP Ib?) polym
orphisms. J Am Coll Cardiol 2003 ;42 :1115-9.
O'Connor F, Shields D, Fitzgerald A, Cannon C, Braunwald E, Fitzgerald D. Genetic variation in glycoprotein IIb/IIIa (GPIIb/IIIa) as a determinant of the responses to an oral GP IIb/IIIa antagonist in patients with unstable coronary syndromes. Blo
od 2001 ;98 :3256-60.
Macchi L, Sorel N, Brabant S, Christiaens L, Mauco G. Relationship between C-5 Kozak, C807T genotypes and expression of glycoproteins Ib? and Ia-IIa at platelet surface : from genotype to phenotype. Thromb Haemost 2002 ;88 :368.
Croft S, Hampton K, Sorrel J, Steeds R, Channer K, Samani N, Daly M. The GPIa C807T dimorphism associated with platelet collagen receptor density is not a risk factor for myocardial infarction. Br J Haematol 1999 ;106 :771-6.
Santoso S, Kunicki TJ, Kroll H, Haberbosch W, Gardemann A. Association of the platelet glycoprotein Ia C807T gene polymorphism with nonfatal infarction in younger patients. Blood 1999 ;93 :2449-53.
Benze G, Heinrich J, Schulte H, Rust S, Nowak-Gottl U, Tataru MC, Kohler E, Assmann G, Junker R. Association of the GP Ia C807T and GP IIIa PlA1/A2 polymorphisms with premature myocardial infarction in men. Eur Heart J 2002 ;23 :325
-30.
Moshfegh K, Wuillemin WA, Redondo M, Lämmle B, Beer JH, Liechti-Gallati S, Meyer BJ. Association of two silent polymorphisms of platelet glycoprotein Ia/IIa receptor with risk of myocardial infarction : a case-control study. Lancet 1999 ;353 :
351-4.
Anvari A, Janisiw M, Turel Z, Huber K, Fisher G, Panzer S. Platelet glycoprotein Ia gene dimorphism ?2-807 in malignant arrythmia in coronary artery disease. Thromb Res 2000 ;98 :281-6.
Von Beckerath N, Koch W, Mehilli J, Bottinger C, Braun S, Schomig A, Kastrati A. Glycoprotein Ia C807T polymorphism and risk of restenosis following coronary stenting. Atherosclerosis 2001 ;156 :463-8.
Mikkelson J, Perola M, Penttila A, Karhunene P. Platelet glycoprotein GP Ib? HPA-2 Met/VNTR B haplotype as a genetic predictor of myocardial infarction an d sudden cardiac death. Circulation 2001 ;104 :876-80.
Murata M, Matsubara Y, Kawano K, Zama T, Aoki N, Yoshino H, Watanabe G, Ishikawa K, Ikeda Y. Coronary artery disease and polymorphisms in a receptor mediating shear stress-dependent platelet activation. Circulation 1997 ;96 : 3281-6.
Douglas H, Michaelides K, Gorog D, Durante-Mangoni E, Ahmed N, Davies G, Tuddenham E. Platelet membrane glcoprotein GP Ib? 5T/C Kozak sequence polymorphism as an independent risk factor for the occurrence of coronary thrombosis. Heart 2002 ;87
:70-4.
Gonzalez-Conjero R, Lozano ML, Rivera J, Corral J, Iniesta J, Moralada J, Vincente V. Polymorphisms of platelet membrane glycoprotein Ib? associated with arterial thrombotic disease. Blood 1998 ;8 :2771-6.
Meisel C, Afshar-Khargan V, Cascorbi I, Laule M, Stangl V, Felix S, Bauman G, Lopez J, Roots I, Stangl K. Role of Kozak sequence polymorphism of platelet glycoprotein Ib? as a risk factor for coronary artery disease and catheter interventions. J A
m Coll Cardiol 2001 ;38 :1023-7.
Frank M, Reiner A, Schwartz S, Kumar P, Pearce R, Arbogast P, Longstreth W, Rosendaal F, Psaty B, Siscovick D. The Kozak sequence polymorphism of platelet glycoprotein Ib and risk of non-fatal myocardial infarction and non-fatal stroke in young women
. Blood 2001 ;97 :875-9.
Croft S, Hampton K, Daly M, Steeds R, Channer K, Samani N. Kozak sequence polymorphism in the platelet GPIb alpha gene is not associated with risk of myocardial infarction. Blood 2000 ;95 :2183-4.
Croft S, Samani N, Teare M, Hampton K, Path M, Steeds R, Channer K, Daly M. Novel platelet membrane glycoprotein VI dimorphism is a risk factor for myocardial infarction. Circulation 2001 ;104 :1459-63.
|







{kind=link}