Introducción
La etiopatogenia y el significado clínico de las lesiones cerebrales de la sustancia
blanca (LSB) no están todavía completamente aclarados. La hipótesis más probable
en la patogenia de estas lesiones es la mediada por un mecanismo
vascular.1 Así, en la mayoría de los estudios, la edad y la
hipertensión arterial (HTA) son los factores que más se relacionan con la presencia
de LSB.2 La elevación de la presión arterial (PA) es capaz de inducir
cambios funcionales y estructurales en las arteriolas terminales que irrigan la
sustancia blanca cerebral, con la consecuente producción de isquemia. Sin
embargo, algunos estudios pusieron de minifiesto que los factores vasculares
explican sólo una parte de la incidencia y variabilidad de las LSB, sugiriendo así la
existencia de otros factores, probablemente genéticos, relacionados con el
desarrollo de LSB. De esta manera, Carmelli y col.3 mostraron en un
estudio realizado en gemelos de sexo masculino (74 monocigotos y 71 dicigotos)
que la susceptibilidad de presentar LSB estaba, en gran parte, determinada
genéticamente (concordancia del 61% en gemelos monocigotos y 38% en
dicigotos). La importancia de la presencia de estas lesiones es que aumenta el
riesgo para el desarrollo posterior de ictus4 y también que cuanto
mayor es la extensión de la LSB el deterioro de la función cognitiva es más
probable.5-7
La relación entre las LSB y los diferentes parámetros clínicos y biológicos asociados
a la HTA no está suficientemente definida. En la población hipertensa, la presencia
de LSB se asocia con la gravedad de las cifras de PA y la ausencia de control de la
PA en hipertensos tratados, puesto en evidencia tanto en estudios
transversales2,5,6,8 como longitudinales,9-11 así
como la redución nocturna de la PA.12,13 Sin embargo, la mayoría
de estudios realizados hasta la fecha se efectuaron en población
anciana5,6,10,11 o en un amplio rango de edades o se incluyeron
pacientes hipertensos con tratamiento antihipertensivo,6,12,13
factores todos ellos que podrían actuar como factores de confusión.
El presente trabajo pretende resumir y contextualizar algunos de los principales
hallazgos provenientes de un estudio de evaluación de los factores relacionados con
la presencia de LSB en una cohorte de pacientes de mediana edad con HTA esencial
que no habían recibido tratamiento previo. Resultados parciales de alguna de dichas
asociaciones se publicaron previamente en forma de originales.14-
17
Pacientes y métodos
Selección de pacientes
Se estudiaron 71 pacientes de ambos sexos (43 hombres, 28 mujeres)
afectados por HTA esencial, grados I o II, nunca tratada y de edades comprendidas
entre 50 y 60 ańos. Los pacientes fueron seleccionados de forma consecutiva de la
Unidad de Hipertensión del Hospital Clínic de Barcelona en su primera visita, a la
que habían sido remitidos para diagnóstico y tratamiento de HTA, si cumplían con
los criterios de inclusión. Todos los pacientes tenían PA sistólica (PAS) ≥ 140
mm Hg, PA diastólica (PAD) ≥ 90 mm Hg o ambas, en al menos tres mediciones
diferentes separadas una semana. El diagnóstico de HTA esencial se consideró si no
se detectaba ninguna causa secundaria de elevación de la PA después de
exploración clínica, analítica y radiológica completa. Los criterios de exclusión
comprendían: diabetes mellitus tipo 2 (glucosa plasmática basal > 110 mg/dl),
estenosis carotídea > 50% determinada por eco Doppler, ingesta enólica > 30
g/día, evidencia clínica de enfermedad cerebrovascular o coronaria, insuficiencia
cardíaca, fibrilación auricular, papiledema e insuficiencia renal (cretinina plasmática
> 1.3 mg/dl).
El estudio fue aprobado por el Comité Etico del Hospital y todos los pacientes dieron
su consentimiento por escrito.
Resonancia magnética cerebral y clasificación de LSB
Las resonancias magnéticas (RM) cerebrales se realizaron con el aparato 1.5
Tesla Siemens Magneton SP (Siemens AG, Erlangen, Alemania). En cada paciente
se realizaron los siguientes cortes: plano axial T1 (técnica spin-eco: tiempo de
repetición [TR] 608 ms, tiempo eco [TE] 14 ms), T2 (TR 2 500 ms, TE 90 ms) y
densidad protón (TR 2 500 ms, TE 15 ms). Asimismo se realizaron imágenes en el
plano sagital con frecuencias cortas (técnica spin-eco TR/TE 608/14 ms) y en el
plano coronal (técnica spin-eco rápida TR/TE 4 600/90 ms). El grosor de los cortes
radiológicos fue de 5 mm. Los datos fueron analizados por dos investigadores que
desconocían los datos clínicos de los pacientes. El diagnóstico de LSB se realizó por
consenso. El protocolo utilizado para la valoración y clasificación de LSB fue el
mismo que el del estudio de Rotterdam.6 La presencia de infartos
lacunares silentes no se consideró en el estudio por resultar marginal en esta
cohorte de pacientes de mediana edad. Los pacientes fueron clasificados en dos
grupos en función de la presencia o ausencia de LSB.
Medida de la presión arterial
La PA clínica se determinó en tres ocasiones, tras un período de descanso de
10 minutos, con un esfigmomanómetro de mercurio. Se consideró la media de las
dos últimas medidas de PA. La monitorización ambulatoria de la PA (MAPA) durante
24 horas se realizó mediante un aparato oscilométrico no invasivo (SpaceLabs
90207; SpaceLabs Inc., Redmond, Washington, EE.UU.). Las determinaciones de PA
se realizaron cada 15 minutos durante todo el período de 24 horas. Dado que
durante la MAPA los pacientes se hallaban hospitalizados, las actividades y horarios
eran similares para todos ellos. Se evaluaron los siguientes parámetros obtenidos
con la MAPA: media de 24 horas, diurna (de 8 h a 23 h) y nocturna (de 23 h a 8 h)
de la PAS, PAD, presión de pulso (PP) y frecuencia cardíaca (FC). El descenso
nocturno de la PA se calculó mediante la diferencia entre la media diurna y la
nocturna. La variabilidad de la PA se determinó mediante las desviaciones estándar
de 24 horas de la PAS y la PAD.
Determinaciones bioquímicas, hormonales y genéticas
Las muestras de sangre se extrajeron por la mańana, en ayunas, después de 1
hora de reposo en cama. Se utilizaron técnicas de laboratorio estándar para la
determinación del perfil bioquímico en suero y orina, así como para la medida de la
actividad de renina plasmática, aldosterona y noradrenalina plasmáticas. La
insulinemia se determinó mediante radioinmunoensayo.
Las muestras para el análisis del ADN procedían de leucocitos de sangre periférica,
según metodología previamente descrita.18 Se determinaron los
siguientes polimorfismos genéticos mediante la técnica de la reacción en cadena de
la polimerasa (PCR): polimorfismo inserción/deleción (I/D) del gen de la enzima de
conversión de la angiotensina (ECA), polimorfismo M235T del gen del
angiotensinógeno y polimorfismo A1166C del gen que codifica el receptor tipo 1 de
la angiotensina (AT1). En función del genotipo, los pacientes fueron
clasificados como II, ID o DD para el polimorfismo del gen de la ECA, como MM, MT
o TT, para el gen del angiotensinógeno, y como AA, AC o CC, para el gen del
receptor tipo 1 de la angiotensina.
Estudio ecocardiográfico
Se realizó ecocardiograma bidimensional en modo M con el paciente en
decúbito lateral izquierdo, después de un descanso de 10 minutos. Se determinaron
los siguientes parámetros de acuerdo con la Sociedad Americana de
Ecocardiografía:19 diámetro telediastólico del ventrículo izquierdo
(DTdVI), grosor de la pared posterior (PP) y grosor del tabique interventricular
(TIV). La masa del VI (MVI) se calculó según el criterio de la convención de
Penn20 y se dividió por la superficie corporal para calcular el índice
de masa del VI (IMVI) en gramos/m2. Se diagnosticó HVI cuando el
IMVI era superior a 110 g/m2 en mujeres y superior a 130
g/m2 en varones.21 El grosor relativo de la pared
(GRP) se obtuvo según la fórmula estándar: 2 x PP/DtdVI.22 Se
consideraron los siguientes patrones geométricos del VI: normal (IMVI normal y
GRp < 0.45), remodelado concéntrico (IMVI normal y GRP ≥ 0.45), hipertrofia
concéntrica (IMVI aumentado y GRP ≥ 0.45) e hipertrofia excéntrica (IMVI
aumentado y GRp < 0.45).
Evaluación neuropsicológica
El test neuropsicológico incluía una estimación del coeficiente intelectual
(vocabulario y cubos de Kohs de la adaptación espańola de la Wechsler Adult
Intelligence Scale [WAIS]),23 pruebas de atención y de
memoria de trabajo (series de dígitos directa e inversa del WAIS-
revisado),24 así como pruebas de valoración de memoria lógica y
visual (revisión de Rusell de la subescala de memoria lógica y subescala de
reproducción visual de la Wechsler Memory Scale).25 Las
pruebas fueron realizadas por un neuropsicólogo. También se realizaron pruebas
para valorar grado de ansiedad o depresión.26
Análisis estadístico
La comparación de los diferentes parámetros entre pacientes hipertensos con
LSB y sin LSB se realizó mediante la prueba de la t de Student y la prueba
U de Mann-Whitney, para variables cuantitativas. La prueba de
χ2 o la prueba exacta de Fisher se utilizaron para las variables
categóricas. Un análisis de regresión logística se utilizó para valorar la asociación
entre la HVI y el patrón geométrico y las LSB, así como para valorar un posible
efecto sinérgico entre los tres polimorfismos genéticos y la presencia de LSB. Los
datos se expresan mediante su media (desviación estándar).
Resultados
En relación con la presencia y la gravedad de LSB en la RM, 43 pacientes no
mostraron LSB (grado 0); 21 pacientes tenían LSB grado I (moderado) y 7
pacientes grado II (grave). Debido al pequeńo número de pacientes que tenían LSB
de grado II y para una mejor identificación de posibles diferencias entre pacientes
con LSB y sin LSB, los pacientes hipertensos se dividieron en dos grupos en función
de la presencia (28 pacientes hipertensos; 39.4%) o ausencia (43 hipertensos;
60.6%) de LSB en la RM cerebral. Las principales características demográficas no
eran diferentes entre ambos grupos, tal y como se muestra en la tabla 1.
Asimismo, tampoco se encontraron diferencias en relación con parámetros
bioquímicos y hormonales (tabla 1).

Presión arterial
En relación con la PA clínica, los pacientes hipertensos con LSB presentaban
cifras de PAS, PAD y PP significativamente superiores a las observadas en los
hipertensos sin LSB, tal y como se muestra en la figura 1. En referencia a los
parámetros obtenidos mediante la MAPA de 24 h también se observaron las mismas
tendencias determinadas en la clínica en relación con la PAS, la PAD y la PP (tabla
2). Con respecto al perfil circadiano y a la variabilidad de la PA no se hallaron
diferencias entre pacientes hipertensos con LSB y sin LSB (tabla 2).


Estudio de los polimorfismos genéticos del sistema renina-
angiotensina
Se determinaron los polimorfismos I/D del gen de la ECA, M235T del gen del
angiotensinógeno y A1166C del gen que codifica el receptor AT1 en
60 pacientes. La distribución de los genotipos en el conjunto de pacientes, así como
las frecuencias de los alelos de los tres polimorfismos se mantuvo en equilibrio de
Hardy-Weinberg. La distribución de los diferentes genotipos entre pacientes con
LSB y sin LSB se muestra en la figura 2. No se observaron diferencias en relación
con los polimorfismos M235T del gen del angiotensinógeno y A1166C del gen que
codifica el receptor AT1. En relación con el polimorfismo I/D del gen
de la ECA, los pacientes con LSB mostraron una significativa (p = 0.022) mayor
frecuencia del genotipo DD (64%) que los pacientes sin lesiones (28.6%), y esta
asociación permaneció significativa tras ajustar para los valores de PAS y PAD (p =
0.049). Asimismo, la proporción del alelo D en los hipertensos con LSB (74%) fue
significativamente superior (p = 0.014) que la observada en pacientes sin lesiones
(51.4%). La odds ratio del genotipo DD para la presencia de LSB fue de
4.44 (intervalo de confianza [IC] del 95%: 1.48-13.32).

Al analizar un posible efecto sinérgico de los tres polimorfismos del sistema renina-
angiotensina y la presencia de LSB no se encontró tal asociación.
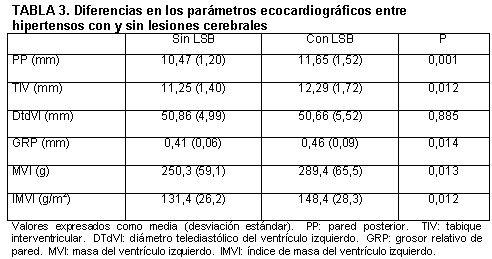
Estudio ecocardiográfico
Se completó el estudio ecocardiográfico en 62 pacientes. Los pacientes
hipertensos con LSB presentaron valores significativamente superiores de PP, TIV,
MVI, IMVI y GRP en comparación con los pacientes sin lesiones, tal y como se
muestra en la tabla 3. La prevalencia de HVI fue significativamente superior (p =
0.01) entre los pacientes con LSB (88.4%) que en los pacientes sin lesiones
(58.3%). La odds ratio de HVI para la presencia de LSB fue de 5.5 (IC
95%: 1.4-31.6). Al analizar la pauta geométrica del ventrículo izquierdo se observó
que los pacientes con LSB presentaban mayor prevalencia de HVI de tipo
concéntrico que los pacientes sin lesiones (54% vs. 11%; p = 0.002). La
odds ratio de hipertrofia concéntrica del VI para la presencia de LSB fue
9.3 (IC 95%: 2.5-34).
Dado que los pacientes con LSB tenían PA superior a los pacientes sin LSB se
realizó un análisis de regresión logística para valorar la relación entre la HVI y el
patrón geométrico del VI con la presencia de LSB, ajustando para los valores de
PAS y PAD. De este modo, la odds ratio de HVI para la presencia de LSB
perdió significación estadística 3.42 (IC 95%: 0.79-14.84). Sin embargo, la
odds ratio de hipertrofia concéntrica permaneció estadísticamente
significativa: 8.22 (IC 95%: 2.06-32.78).
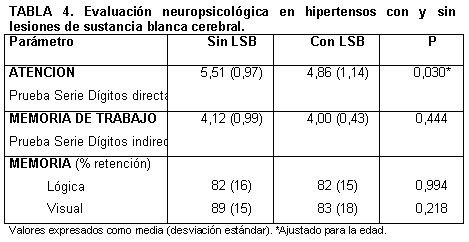
Evaluación neuropsicológica
Se completó el estudio neuropsicológico en 60 pacientes. Los pacientes con
LSB mostraron una puntuación significativamente peor en la prueba de las series de
dígitos directa, medida estándar de la capacidad de atención, que los pacientes sin
lesiones (4.86 ± 1.14 vs. 5.51 ± 0.97; p = 0.027). Esta
diferencia permaneció significativa tras ajustarla por la edad (p = 0.030). No se
hallaron diferencias en la memoria de trabajo ni en la memoria visual y lógica
(tabla 4). No se encontraron diferencias entre ambos grupos en relación con
coeficiente intelectual, nivel de educación o nivel de ansiedad/depresión.
Discusión
Los resultados del presente estudio muestran que la prevalencia de LSB en un
grupo de pacientes hipertensos de mediana edad es de 39.4%. La presencia de LSB
se relaciona con la gravedad de la HTA y no con el perfil circadiano ni la variabilidad
de la PA. Asimismo, la existencia de estas lesiones se asocia con deterioro leve de la
capacidad de atención y con mayor presencia de hipertrofia concéntrica del VI y del
genotipo DD del gen que codifica la ECA.
La prevalencia de LSB en los distintos estudios realizados hasta la fecha oscila entre
5% y 55%.5,6,8,27 Esta gran discordancia está ocasionada,
probablemente, por las caracteristícas de los pacientes incluidos en cada estudio,
así como por el criterio utilizado para la valoración de la presencia de LSB. La
mayoría de estudios incluyeron una mezcla de sujetos normotensos e hipertensos o
sólo individuos de más de 65 ańos.5,6 Asimismo, en algunos
estudios se consideró el diagnóstico de HTA cuando las cifras de PA eran > 160/95
mm Hg,6 o bien se incluían pacientes hipertensos tratados
farmacológicamente,6,27 ańadiendo así mayor confusión en relación
con la posible influencia del tratamiento antihipertensivo o del control de la PA en la
incidencia de LSB.
Los resultados del presente trabajo confirman lo observado en anteriores estudios
con respecto a la relación entre la gravedad de las cifras de PA y la existencia de
LSB.2,9-11 En efecto, los pacientes hipertensos con PA clínica más
elevada, tanto sistólica como diastólica, muestran mayor presencia de LSB. Otra de
las características que otorga más importancia a la relación entre la presencia de
LSB y la gravedad de la HTA es la confirmación de dicha relación con la MAPA. Es
conocido el hecho de que la PA obtenida durante un período de 24 horas se
correlaciona mejor con otros marcadores de lesión de órgano diana como la HVI o
la microalbuminuria. El presente estudio enfatiza este hecho al encontrar una
asociación entre las cifras de PA obtenidas por MAPA y la presencia de LSB.
Otro de los datos interesantes del estudio es la asociación entre la elevación de la
presión de pulso y la presencia de LSB. Para algunos autores, la PP es un mejor
predictor de riesgo cardiovascular que las propias cifras de PAS y PAD por separado
y, en los últimos ańos, surgieron datos muy convincentes para considerar la PP
como un factor de riesgo independiente para la morbimortalidad
cardiovascular.28 Hasta la fecha sólo un estudio había relacionado la
PP en la consulta con la gravedad de las LSB en una muestra de individuos de
mayor edad (55-72 ańos).29
El perfil circadiano de la PA no se asoció con la existencia de LSB en el presente
trabajo. El estudio de Shimada y col.12,13 sí mostró asociación
entre la presencia de lesiones silentes cerebrovasculares (que incluían tanto
lacunares como LSB) y el ritmo circadiano. Así, los pacientes con un perfil non-
dipper y los extreme-dipper presentaban más lesiones cerebrales que
los pacientes con un perfil dipper. Esta discordancia podría estar
relacionada por las características de las muestras, como se mencionó
anteriormente. En este sentido es preciso comentar que los individuos del estudio
de Shimada y col.12,13 tenían más edad y algunos de ellos recibían
tratamiento antihipertensivo. Asimismo, en el estudio de Shimada y col. las lecturas
de PA durante la monitorización se realizaban cada 30 minutos. En el presente
trabajo las determinaciones de la PA se efectuaban cada 15 minutos para obtener
mayor número de medidas y mejor valoración del perfil circadiano.
Al igual que el perfil circadiano, la variabilidad de la PA a largo plazo no se relacionó
con la presencia de LSB en el presente estudio. Goldstein y col.30
mostraron asociación entre la variabilidad de la PAS, sólo durante el período diurno
y la existencia de LSB. Por el contrario, el estudio de Shimada y
col.,12,13 realizado en 73 individuos normotensos e hipertensos de
edad avanzadano encontró relación entre la variabilidad de la PA y la presencia
tanto de infartos lacunares como de LSB.
La prevalencia de HVI en hipertensos con LSB es superior a la observada en
pacientes sin lesiones. No obstante, al realizar el ajuste estadístico, dado que los
hipertensos con LSB presentan cifras de PA significativamente mayores, este dato
no es independiente de los valores de la PA. Algunos estudios previos sobre la
relación entre la presencia de HVI y LSB son contradictorios, ciertos estudios sí
mostraron tal asociación5,31 y otros no.27,32 La
heterogeneidad de los resultados también podría estar en consonancia con la
heterogeneidad de las muestras en los diferentes estudios (edad
avanzada,5,27,32 antecedentes de cardiopatía
previa,5 tratamiento antihipertensivo
concomitante27,31,32) o con el método utilizado para la evaluación
de la HVI. En el presente trabajo se incluyeron pacientes hipertensos que nunca
habían recibido tratamiento antihipertensivo. De esta manera, se intentaba evitar la
posible influencia que el tratamiento puede ejercer en la aparición tanto de
LSB8,10 como de HVI.33 Además se utilizó
ecocardiografía, método con mayor sensibilidad, y no el electrocardiograma para la
evaluación de la HVI.34
Al analizar el patrón geométrico de la MVI se halló que los pacientes hipertensos
con LSB muestran mayor prevalencia de hipertrofia concéntrica del VI que los
pacientes sin lesiones, independientemente de las cifras de PA. Hasta la fecha se
había relacionado la hipertrofia concéntrica con mayor repercusión de órgano diana
a nivel renal y retiniano,35,36 pero no existían estudios que
hubieran relacionado este tipo de hipertrofia con la presencia de LSB.
En el presente estudio observamos que la presencia de LSB está asociada con el
polimorfismo I/D del gen de la ECA en pacientes hipertensos de mediana edad. De
este modo, los pacientes hipertensos con LSB muestran mayor prevalencia del
genotipo DD del gen de la ECA que los hipertensos sin lesiones,
independientemente de las cifras de PA. Del mismo modo, los hipertensos con
lesiones presentan mayor frecuencia del alelo D que los pacientes sin lesiones.
Previamente, un estudio realizado en un grupo de individuos ancianos afectados por
demencia encontró asociación entre el genotipo DD del gen de la ECA y la presencia
de LSB sólo en aquellos pacientes con antecedentes de ictus.37
Al analizar otros polimorfismos relacionados con el sistema renina-angiotensina,
como el gen M235T del angiotensinógeno o el gen A1166C del receptor tipo 1 de la
angiotensina II, no hallamos ninguna relación con la presencia de LSB. Por el
contrario, Schmidt y col.38 sí observaron asociación entre el
genotipo TT del gen del angiotensinógeno y la presencia de LSB. Es preciso
destacar que en el estudio de Schmidt y col.38 la muestra estaba
compuesta por individuos normotensos e hipertensos de edad más avanzada (50 a
75 ańos) y que 37% de ellos tenían antecedentes de enfermedad cardíaca, factores
que pueden influir la presencia de LSB. Por el contrario, en el estudio de Takami y
col.39 tampoco se demostró asociación entre el polimorfismo del gen
del angiotensinógeno y la presencia de lesiones periventriculares en un grupo de
individuos normotensos e hipertensos. Por lo que respecta a la asociación entre el
gen A1166C del receptor tipo 1 de la angiotensina II y la presencia de LSB, en un
estudio39 previo se encontró que individuos de mediana edad (51 a
60 ańos) normotensos e hipertensos con el genotipo AC mostraban mayor grado de
lesiones periventriculares que los individuos con el genotipo AA, mientras que esta
asociación no existía en los individuos de mayor edad (61 a 70 ańos). Es preciso
destacar que en este estudio no se incluyeron individuos con el genotipo CC.
Tampoco encontramos un posible efecto sinérgico entre estos tres genes y la
presencia de LSB. Hasta la fecha sólo un estudio40 detectó
interacción positiva entre el genotipo TT del gen del angiotensinógeno y el genotipo
DD del gen de la ECA con la presencia de ictus. Sin embargo, no se realizaron
estudios que valoren la presencia o ausencia de un efecto sinérgico entre estos tres
genes y las LSB.
En relación con la evaluación neuropsicológica, hemos observado que los pacientes
con LSB mostraban una puntuación significativamente peor en la prueba de la serie
de dígitos directa, que es una medida de la capacidad de atención. La relación entre
la presencia de LSB y un deterioro de la función cognitiva es un dato ya conocido,
tanto en estudios transversales como longitudinales.7,41 Sin
embargo, el presente estudio, a diferencia de otros, se realizó en pacientes de
mediana edad para intentar valorar posibles asociaciones entre LSB y deterioro
cognitivo en etapas tempranas. El presente estudio sólo encuentra diferencias en la
capacidad básica de atención. Se desconoce todavía si la alteración de la capacidad
de atención es una característica típica asociada con la presencia de LSB, y si es el
área que primero se afecta como consecuencia de la existencia de LSB. En un
estudio realizado también en pacientes hipertensos, Schmidt y col.42
no encontraron diferencias en las series de dígitos directa e indirecta pero sí en
pruebas que valoraban concentración y velocidad de procesamiento mental. Al igual
que en el presente estudio, Schmidt y col. tampoco encontraron diferencias en las
pruebas de memoria. Las diferencias entre los estudios podrían ser explicadas por
las distintas carcterísticas de las muestras: la población hipertensa de Schmidt y
col. era mayor y el 50% estaba en tratamiento antihipertensivo. Van Swieten y
col.43 encontraron en ancianos hipertensos con LSB (n = 10) peores
puntuaciones en las pruebas de concentración, procesamiento y de memoria visual
que ancianos hipertensos sin LSB (n = 24) y no hallaron diferencias en las series de
dígitos. En ese estudio, algunos pacientes tenían antecedentes de enfermedad
cardiovascular previa y se definía la HTA con valores > 160/95 mm Hg.
A pesar de las posibles limitaciones del presente estudio, como el tamańo de la
muestra o el diseńo transversal, la inclusión de un grupo homogéneo de pacientes
de mediana edad, asintomáticos, sin antecedentes de enfermedad cardiovascular y
sin otros factores de riesgo hacen que los factores de confusión se limiten y que las
relaciones halladas tengan más interés.
En conclusión, la presencia de LSB en pacientes asintomáticos de mediana edad
afectados por HTA esencial se relaciona con la gravedad de la elevación de la PA,
así como con deterioro cognitivo incipiente y con mayor afectación de órgano diana,
en forma de HVI de tipo concéntrico. Asimismo, la aparición de LSB en la HTA
esencial puede estar relacionada con determinados factores genéticos, como el
genotipo DD del gen de la ECA.
Los autores no manifiestan conflictos.
BIBLIOGRAFÍA
-
Van Swieten JC, Van den Hout JH, Van Ketel BA, Hijdra A, Wokke JH, Van Gijn J.
Periventricular lesions in the white matter on magnetic resonance imaging in the
elderly: a morphometric correlation with arteriolosclerosis and dilated perivascular
spaces. Brain 1991;114:761-774.
-
Pantoni L, García JH. The significance of cerebral white matter abnormalities 100
years after Binswanger’s report. A review. Stroke 1995;26:1293-1301.
-
Carmelli D, DeCarli C, Swan GE, Jack LM, Reed T, Wolf PA, Miller BL. Evidence
for genetic variance in white matter hyperintensity volume in normal elderly male
twins. Stroke 1998;29:1177-1181.
-
Van Swieten JC, Kapelle LJ, Algra A, Van Latum JC, Koudstaal PJ, Van Gijn J.
Hipodensity of the cerebral white matter in patients with transient ischemic attack
or minor stroke: influence on the rate of subsequent stroke. Duth TIA Trial Study
Group. Ann Neurol 1992;32:177-183.
-
Longstreth WT, Manolio TA, Arnold A, Burke GL, Bryan N, Jungreis ChA, et al.
Clinical correlates of white matter findings on cranial magnetic resonance imaging of
3301 elderly people: The Cardiovascular Health Study. Stroke 1996;27:1274-1282.
-
Breteler MMB, Van Swieten JC, Bots ML, Grobbee DE, Claus JJ, Van den Hout
JHW, et al. Cerebral white matter lesions, vascular risk factors, and cognitive function
in a population-based study: The Rotterdam Study. Neurology 1994;44:1246-1252.
-
De Groot JC, de Leeuw FE, Oudkerk M, Van Gijn J, Hofman A, Jolles J, Breteler
MMB. Periventricular cerebral white matter lesions predict rate of cognitive decline.
Ann Neurol 2002;52:335-341.
-
Liao D, Cooper L, Cai J, Toole JF, Bryan NR, Hutchinson RG, et al. Presence and
severity of cerebral white matter lesions and hypertension, its treatment, and its
control: The ARIC Study. Stroke 1996;27:2262-2270.
-
Schmidt R, Fazekas F, Kapeller P, Schmidt H, Hartung HP. MRI white matter
hyperintensities. Three-year follow-up of the Austrian Stroke Prevention Study.
Neurology 1999;53:132-139.
-
Dufouil C, De Kersaint-Gilly A, Besançon V, Levy C, Auffray E, Brunnereau L et al.
Longitudinal study of blood pressure and white matter hyperintensities. The EVA MRI
Cohort. Neurology 2001;56:921-926.
-
De Leeuw FE, De Groot JC, Oudkerk M, Witteman JCM, Hofman A, Van Gijn J et
al. Hypertension and cerebral white matter lesions in a prospective cohort study.
Brain 2002;125:765-772.
-
Shimada K, Kawamoto A, Matsubayashi K, Nishinaga M, Kimura S, Ozawa T.
Diurnal blood pressure variations and silent cerebrovascular damage in elderly
patients with hypertension. J Hypertens 1992;10:875-878.
-
Kario K, Matsuo T, Kobayashi H, Imiya M, Matsuo M, Shimada K. Nocturnal fall
of blood pressure and silent cerebrovascular damage in elderly hypertensive
patients. Advanced silent cerebrovascular damage in extreme dippers. Hypertension
1996;27:130-135.
-
Sierra C, Coca A, Gómez-Angelats E, Poch E, Sobrino J, De la Sierra A. Renin-
angiotensin system genetic polymorphisms and cerebral white matter lesions in
essential hypertension. Hypertension 2002;39:343-347.
-
Sierra C, De la Sierra A, Mercader J, Gómez-Angelats E, Urbano-Márquez A,
Coca A. Silent cerebral white matter lesions in middle-aged essential hypertensive
patients. J Hypertens 2002;20:519-524.
-
Sierra C, De la Sierra A, Paré JC, Gómez-Angelats E, Coca A. Correlation
between silent cerebral white matter lesions and left ventricular mass and geometry
in essential hypertension. Am J Hypertens 2002;15:507-512.
-
Sierra C, De la Sierra A, Salamero M, Gómez-Angelats E, Sobrino J, Coca A.
Silent cerebral white matter lesions and cognitive function in middle-aged essential
hypertensive patients. Am J Hy pertens 2004;17:529-534.
-
Leadon SA, Cerutti PA. A rapid and mild procedure for the isolation of DNA from
mammalian cells. Anal Biochem 1982;120:282-288.
-
Sahn DJ, De Maria A, Kisslo J. Weyman A. The Committee on M-Mode
Standardization of the American Society of Echocardiography: recommendations
regarding quantitation in M-mode echocardiography: results of a survey of
echocardiographic measurements. Circulation 1978;58:1072-1083.
-
Devereux RB, Reichek N. Echocardiographic determination of left ventricular
mass in man: anatomic validation of the method. Circulation 1977;55:613-618.
-
Devereux RB, Casale PN, Kligfield P, Eisenberg RR, Miller D, Campo E, Alonso
DR. Performance of primary and derived M-mode echocardiographic measurements
for detection of left ventricular hypertrophy in necropsied subjects and in patients
with systemic hypertension, mitral regurgitation and dilated cardiomiopathy. Am J
Cardiol 1986;57:1388-1393.
-
Reichek N, Devereux RB. Reliable estimation of peak left ventricular systolic
pressure by M-mode echocardiographic-determined end-diastolic relative wall
thickness: identification of severe valvular aortic stenosis in adult patients. Am
Heart J 1982;103:202-203.
-
Wechsler D. WAIS. Escala de inteligencia de Wechsler para adultos. New York.
The Psychological Corporation. Tea Ediciones. Madrid. 1990.
-
Kaplan E, Fein D, Morris R. Delis DC. WAIS-R as a neuropsychological
instrument. New York. The psychological corporation. 1991.
-
Rusell EW. A multiple scoring method for the assessment of complex memory
functions. J Consult Clin Psycol 1975;43:800-809.
-
Zigmond AS, Snaith RP. The Hospital Anxiety and Depression Scale. Acta
Psychiatr Scand 1983;67:361-370.
-
Shimada K, Kawamoto A, Matsubayashi K, Ozawa T. Silent cerebrovascular
disease in the elderly. Correlation with ambulatory pressure. Hypertension
1990;16:692-699.
-
Benetos A, Rudnichi A, Safar M, Guize L. Pulse pressure and cardiovascular
mortality in normotensive and hypertensive subjects. Hypertension 1998;32:560-
564.
-
Liao D, Cooper L, Cai J, Toole J, Bryan N, Burke G, et al. The prevalence and
severity of white matter lesions, their relationship with age, ethnicity, gender, and
cardiovascular disease risk factors: the ARIC study. Neuroepidemiology
1997;16:149-162.
-
Goldstein IB, Bartzokis G, Hance DB, Shapiro D. Relationship between blood
pressure and subcortical lesions in healthy elderly people. Stroke 1998;29:765-772.
-
Lindgren A, Roijer A, Rudling O, Norrving B, Larsson E, Eskilsson J, et al.
Cerebral lesions on magnetic resonance imaging,heart disease, and vascular risk
factors in subjects without stroke. A population-based study. Stroke 1994;25:929-
934.
-
Kario K, Matsuo T, Kobayashi H, Hoshide S, Shimada K. Hyperinsulinemia and
hemostatic abnormalities are associated with silent lacunar cerebral infarcts in
elderly hypertensive subjects. J Am Coll Cardiol 2001;37:871-877.
-
Schmieder RE, Martus P, Klingbeil A. Reversal of left ventricular hypertrophy in
essential hypertension. JAMA 1996;275:1507-1513.
-
Devereux RB, Reichek N. Echocardiographic determination of left ventricular
mass in man: anatomic validation of the method. Circulation 1977;55:613-618.
-
Shigematsu Y, Hamada M, Ohtsuka T, Hashida H, Ikeda S, Kuwahara T, et al.
Left ventricular geometry as an independent predictor for extracardiac target organ
damage in essential hypertension. Am J Hypertens 1998;11:1171-1177.
-
Pontremolli R, Ravera M, Bezante GP, Viazzi F, Nicolella C, Berruti V, et al. Left
ventricular geometry and function in patients with essential hypertension and
microalbuminuria. J Hypertens 1999;17:993-1000.
-
Amar K, MacGowan S, Wilcock G, Lewis T, Scott M. Are genetic factors
important in the etiology of leukoaraiosis? Results from a memory clinic population.
Int J Geriatric Psychiatry 1998;13:585-590.
-
Schmidt R, Schmidt H, Fazekas F, Launer LJ, Niederkorn K, Kapeller P, et al.
Angiotensinogen polymorphism M235T, carotid atherosclerosis, and small-vessel
disease-related cerebral abnormalities. Hypertension 2001;38:110-115.
-
Takami S, Imai Y, Katsuya T, Ohkubo T, Tsuji I, Nagai K, et al. Gene
polymorphism of the renin-angiotensin system associates with risk for lacunar
infarction. Am J Hypertens 2000 ;13 :121-127.
-
Nakata Y, Katsuya T, Rakugi H, Takami S, Sato N, Kamide K, et al.
Polymorphism of angiotensin converting enzyme, angiotensinogen, and
apolipoprotein E genes in a Japanese population with cerebrovascular disease. Am J
Hypertens 1997;10:1391-1395.
-
De Groot JC, de Leeuw FE, Oudkerk M, Van Gijn J, Hofman A, Jolles J, Breteler
MMB. Cerebral white matter lesions and cognitive function: The Rotterdam Scan
Study. Ann Neurol 2000;47:145-151.
-
Schmidt R, Fazekas F, Koch M, Kapeller P, Augustin M, Offenbacher H, Fazekas
G, Lechner H. Magnetic resonance imaging cerebral abnormalities and
neuropsychologic test performance in elderly hypertensive subjects. Arch Neurol
1995;52:905-910.
-
Van Swieten JC, Geyskes GG, Derix MMA, Peeck BM, Ramos LMP, Van Latum
JC, Van Gijn J. Hypertension in the elderly is associated with white matter lesions
and cognitive decline. Ann Neurol 1991;30:825-830.
|



