

Volumen 10, Número 4, Julio 2004
![]()
|
|
|
|
|
|
||
|
Expertos Invitados |
|
|
|
|
|
Columnista
Experto de SIIC Dra. Gemma Llaverias Profesora Titular. Farmacología |
Introducción Tabla 1. Genes diferencialmente expresados en macrófagos THP-1 tratados con oxLDL. La tabla muestra el nombre y el número de Genbank de los genes que resultaron diferencialmente expresados en células expuestas a LDL oxidadas (tratadas) respecto de las células tratadas con vehículo (control). La tercera columna corresponde a la expresión relativa de cada gen, calculada como la relación entre la intensidad ajustada de las células tratadas respecto de las controesl. La última columna indica la categoría funcional a la cual pertenece cada gen. Los resultados son representativos de tres experimentos independientes realizados con diferentes lotes de células y lipoproteínas.  Figura 1. Análisis de los niveles de ARNm de FABP4, CD18 y MMP9 mediante RT-PCR de macrófagos THP-1 tratados con LDL oxidadas durante 24 horas. Se muestra una autorradiografía representativa y la cuantificación de dichos niveles de ARNm normalizados respecto de los del gen de la tubulina 1 (TUBA1). Los resultados se expresan en unidades arbitrarias como la media ± desviación estándar de tres experimentos independientes realizados con diferentes lotes de células y de lipoproteínas. * p < 0.05.Paralelamente se procedió a determinar el efecto del tratamiento de dichas células con avasimibe. El tratamiento con este fármaco produjo una reducción en la expresión de 13 genes, mientras que un total de 4 resultaron inducidos (Tabla 2). El análisis de los niveles de ARNm de FABP4 por RT-PCR confirmó que el avasimibe produce una reducción del 38% en la expresión de dicho gen (Figura 2). Tabla II Tabla 2. Genes diferencialmente expresados en macrófagos THP-1 tratados con avasimibe. La tabla muestra el nombre y el número de Genbank de los genes que resultaron diferencialmente expresados en células expuestas a avasimibe (2 μM durante 24 horas) en presencia de LDL oxidadas (tratadas) respecto de las células incubadas únicamente con estas LDL (control). La tercera columna corresponde a la expresión relativa de cada gen, calculada como la relación entre la intensidad ajustada de las células tratadas respecto a las controles. La última columna indica la categoría funcional a la cual pertenece cada gen. Los resultados son representativos de tres experimentos independientes realizados con diferentes lotes de células y lipoproteínas. 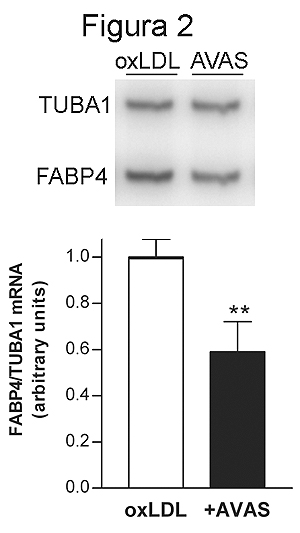 Figura 2. Análisis de los niveles de ARNm de FABP4 mediante RT-PCR de macrófagos THP-1 tratados con avasimibe (2 μM durante 24 horas) en presencia de LDL oxidadas respecto de las mismas células incubadas únicamente con estas LDL. Se muestra una autoradiografía representativa y la cuantificación de dichos niveles de ARNm normalizados respecto de los del gen de la tubulina 1 (TUBA1). Los resultados se expresan en unidades arbitrarias como la media ± desviación estándar de tres experimentos independientes realizados con diferentes lotes de células y de lipoproteínas. ** p < 0.01.Discusión El monocito/macrófago es una célula clave en el desarrollo del proceso aterosclerótico.12 En sus fases iniciales, la captación de lipoproteínas oxidadas conduce a la formación de células espumosas en la pared arterial. Uno de los objetivos del presente estudio era investigar el proceso de aterogénesis desde el punto de vista de la regulación de la expresión génica. Para ello, se realizaron los experimentos en monocitos THP-1, diferenciados a macrófagos tras exposición a ésteres de forbol, un tipo de células ampliamente utilizado como modelo de formación de células espumosas.13 El tratamiento de dichas células con LDL oxidadas produjo modificaciones en la expresión de una serie de genes, cuya función podría estar relacionada con la aterosclerosis. Cuando estos genes se clasificaron según su localización celular y su función, se observó que principalmente se producía una inhibición de la expresión de genes que codifican proteínas localizadas en el retículo endoplasmático o dirigidas hacia la zona extracelular (proteínas de membrana o secretadas al medio). Este patrón coincide con el descrito por Andersson y col.14 en un modelo similar al nuestro. Entre los genes que resultan inducidos tras la exposición a LDL oxidadas, podemos destacar la glucoproteína de membrana CD68. Se trata de un receptor scavenger de clase D, que se expresa únicamente en macrófagos y células dendríticas, capaz de unir e internalizar LDL oxidadas.15 Recientemente se ha propuesto que CD68 desempeńa un papel crucial en la progresión de macrófagos a células espumosas.16 Otro de los genes regulados por LDL oxidadas es la fatty acid binding protein 4 (FABP 4), también denominada ap2. Aunque inicialmente fue identificada en adipocitos, la FABP4 se expresa también en macrófagos.17 Su expresión resulta incrementada 17 veces, respecto de macrófagos control, según los resultados obtenidos de los arrays de ADNc, y 8.4 veces en los estudios realizados por RT-PCR (p < 0.05). Nuestros resultados coinciden con los de Fu y col.,18 que sugieren que el mecanismo por el cual las LDL modificadas inducen la expresión de FABP4 en estas células está relacionado con la activación de la vía de NF-κB. Se ha visto que FABP4 se halla altamente expresada en macrófagos de lesiones ateroscleróticas de conejos hipercolesterolémicos.19 Nuestros resultados demuestran que la adición de avasimibe junto con las LDL oxidadas atenúa la inducción de FABP4 producida por dichas lipoproteínas. Así, la relación de la expresión de FABP4 en células tratadas con LDL oxidadas y avasimibe, respecto de células tratadas con LDL oxidadas en ausencia de fármaco es de 0.18 y 0.62 (p < 0.01), según los resultados de los arrays y de los estudios de RT-PCR, respectivamente. La ausencia de FABP4 en el macrófago protege a los ratones apoE-/- del desarrollo de aterosclerosis.20 Por tanto, la inhibición de la expresión de FABP4 en el macrófago puede representar un nuevo mecanismo de acción antiaterosclerótica directa de avasimibe, que dificultaría la acumulación de colesterol en el macrófago. Otro gen cuya expresión resulta reducida en macrófagos expuestos a avasimibe y LDL oxidadas es la HDL binding protein (HBP) o vigilin. Es una proteína de membrana, capaz de unirse a las HDL, aunque no parece comportarse como un receptor de membrana típico.21 Aunque su función no está clara, se ha propuesto que participa en el metabolismo intracelular de esteroles. Estudios en conejos han mostrado que HBP se expresa en macrófagos cargados de lípidos, colocalizada con apo E.22 Se ha propuesto que factores similares pueden afectar la expresión de ambos genes en macrófagos. De hecho, nuestros resultados muestran una reducción simultánea en la expresión de HBP y de apo E. Así, la relación de la expresión es de 0.45 y 0.35 para HBP y apo E, respectivamente. El hecho de que HBP se halle expresada en macrófagos de lesiones ateroscleróticas indica que su inhibición tras el tratamiento con avasimibe podría constituir un efecto beneficioso, hasta el momento no descrito para ningún otro fármaco, respecto del desarollo de la aterosclerosis. Por lo que respecta a la apo E, en general se considera una proteína antiaterogénica. En concreto, la apo E secretada por los macrófagos actúa como un aceptor de colesterol libre que es liberado hacia el exterior de la célula,23 facilitando el transporte inverso de colesterol. Este papel protector se ha confirmado en estudios en ratones deficientes en apo E, pero cuyos macrófagos sí expresan esta apolipoproteína.24 Estos animales, pese a que presentan niveles circulantes de apo E muy bajos, muestran resistencia al desarrollo de aterosclerosis. La expresión de apo E en el macrófago se halla regulada por los niveles de colesterol.25 Otros autores observaron reducciones en la expresión de apo E en macrófagos tratados con estatinas y atribuyeron este efecto a la reducción del contenido de colesterol intracelular.26 En nuestro caso, el efecto del avasimibe consiste en inhibir la esterificación de colesterol, pero no reduce los niveles de colesterol libre en el interior del macrófago,8 por lo que este mecanismo no explicaría la reducción en los niveles de ARNm de apo E. Puede resultar paradójico que fármacos potencialmente antiaterogénicos como estatinas o inhibidores de la ACAT reduzcan la expresión de una proteína antiaterogénica. Sin embargo, en nuestras condiciones de trabajo, en las que los macrófagos están expuestos a concentraciones muy elevadas de lipoproteínas, la reducción en la secreción de apo E podría representar una respuesta adaptativa destinada a reducir la captación de éstas. Por último, uno de los procesos implicados directamente en la progresión y desarrollo de la placa de ateroma es la angiogénesis.27 La degradación de la matriz extracelular (ECM) resulta clave en el proceso angiogénico y es llevada a cabo por sistemas proteolíticos como el sistema fibrinolítico (plasminógeno/plasmina) y las metaloproteinasas (MMP).28 Existen puntos de contacto entre ambos sistemas: el activador tisular de plasminógeno (tPA) convierte el plasminógeno en plasmina, la cual puede activar las MMP latentes. Estas son una familia de enzimas encargadas de degradar la ECM, lo cual predispone la placa de ateroma a la ruptura. Todas las células vasculares pueden secretar MMP y, en el caso de los macrófagos, la mayoritaria es la MMP- 9.29 La actividad de estos enzimas está regulada no sólo a nivel de expresión génica sino también por los inhibidores tisulares de metaloproteinasas (TIMP), principalmente TIMP-1.30 En el presente estudio se demuestra que el tratamiento con avasimibe reduce la expresión de diversos componentes de estos sistemas, como MMP-9, tPA y TIMP-1. La menor expresión de MMP-9 y de tPA sugiere que el avasimibe podría actuar inhibiendo la angiogénesis, lo cual sería beneficioso en cuanto a limitar el desarrollo de la placa de ateroma. Además, la inhibición d la MMP-9 reduciría la degradación de la ECM, ralentizando el proceso de aterogénesis y aumentando la estabilidad de las placas de ateroma ya formadas. Sin embargo, el efecto final de avasimibe sobre la actividad MMP-9 puede resultar contrarrestado por la reducción en la expresión de su inhibidor TIMP-1. En conclusión, la utilización de técnicas de array de ADNc ha pemitido identificar nuevos genes potencialmente implicados en el desarrollo de la aterosclerosis, cuya expresión resulta modulada en macrófagos humanos expuestos a LDL oxidadas y al inhibidor de la ACAT avasimibe. Entre los efectos más interesantes obtenidos hasta el momento, se destaca la marcada inducción de la FABP4 por LDL oxidadas, que resulta revertida, al menos en parte, por el tratamiento con avasimibe. La FABP4 presenta altos niveles de expresión en macrófagos de lesiones ateroscleróticas, mientras que la falta de expresión de esta proteína protege respecto del desarrollo de lesiones en modelos animales susceptibles.
|
|
|
|
|
 |
Columnista
Experto de SIIC Dr. Aldo D. Mottino Profesor Asociado Area Fisiología, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario. Investigador principal, Instituto de Fisiología Experimental, CONICET. Fisiopatología-Gastroenterología-Metabolismo y transporte de xenobióticos. |
|
|
|
Trabajos Distinguidos, Clínica Médica , integra el Programa SIIC de Educación Médica Continuada |
![]()
Bienvenidos
a siicsalud
Acerca de
SIIC Estructura de SIIC
Sociedad Iberoamericana de Información
Científica (SIIC)
Av. Belgrano 430, (C1092AAR), Buenos Aires, Argentina
atencionallector@siicsalud.com;
Tel: +54 11 4342-4901; Fax: +54 11 4331-3305.
Casilla de Correo 2568, (C1000WAZ) Correo Central, Buenos
Aires.
Copyright siicsalud© 1997-2004, Sociedad Iberoamericana de Información Científica (SIIC)


