Cada ańo fallecen cerca de 60 000 hombres y mujeres por cáncer colorrectal en los Estados Unidos, por lo cual es la segunda causa principal de muerte por cáncer. El estudio Global Burden of Disease estimó que en el ańo 2000 el cáncer de colon fue responsable del fallecimiento de cerca de medio millón de personas en el mundo.1 Debido a estas cifras, esta neoplasia ha sido el centro de atención de intensas investigaciones. La investigación clínica básica realizada durante tres décadas ha arrojado ideas extraordinarias en cuanto a la base genética del cáncer colorrectal que ya han comenzado a tener influencia en el cuidado y atención de los pacientes. En los tres ańos que han transcurrido desde que revisamos este aspecto para Hematology/Oncology Clinics of North America,2 hemos observado diversos avances, como la instauración de terapias de quimioprevención aprobadas para pacientes con poliposis adenomatosa familiar (PAF),3-5 el reconocimiento de un nuevo síndrome de cáncer de colon familiar –la poliposis adenomatosa MYH (PAM)–,6-9 la adopción de nuevas modalidades de pesquisa para el cáncer de colon10-11 y el empleo de terapéuticas biológicas novedosas dirigidas hacia esta neoplasia.12 Este trabajo recopila los conceptos principales de nuestro estudio previo e informa sobre los avances importantes ocurridos desde su publicación.
Conceptos de tumorigénesis colorrectal
Secuencia adenoma-carcinoma
La mayoría de los cánceres colorrectales de los seres humanos surgen de masas displásicas pero no malignas en el colon, llamadas adenomas.2,13- 16 Los pólipos adenomatosos se forman en el colon cuando los mecanismos que regulan la renovación del epitelio se interrumpen o desorganizan.2 En los adenomas, estos mecanismos normales se van interrumpiendo progresivamente a medida que su tamańo y el grado de displasia se incrementan. La secuencia adenoma-carcinoma describe esta progresión desde la mucosa normal hasta el carcinoma invasivo y está bien avalada por numerosos estudios patológicos, epidemiológicos, clínicos observacionales y en animales.2,13-16
Los focos crípticos aberrantes (FCA) son lesiones microscópicas que se cree son el paso intermedio entre la mucosa colónica normal y el pólipo adenomatoso.17-19 La mayoría de los FCA son hiperplásicos (65% a 95%), aunque en una proporción significativa son displásicos y similares a los adenomas.20,21 El análisis genético de los FCA identificó mutaciones que se observan típicamente en los pólipos adenomatosos.17,20,22- 28 Debido a que se considera que los FCA son las lesiones precursoras más tempranas en la progresión hacia el cáncer de colon se los está empleando como biomarcadores tempranos para neoplasia, tanto en estudios en animales como en seres humanos.
La carcinogénesis colónica es un proceso progresivo
Las herramientas de la biología molecular ayudaron a establecer que la carcinogénesis colónica es un proceso escalonado.2,14,29,30 En cada paso del proceso se adquieren mutaciones somáticas o epigenéticas en el ADN celular. Es más probable que los fenómenos de mutación sean silentes, o que sean perjudiciales para la supervivencia de la célula, en vez de promover la neoplasia.
Sin embargo, algunas mutaciones activan vías promotoras del crecimiento (por ejemplo oncogenes) o inactivan supresores tumorales y vías apoptóticas.31,32 Estas clases de mutaciones aportan una ventaja en la supervivencia, lo que conduce a la expansión clonal y a la formación de una masa.33,34 En una revisión fundamental sobre las características sobresalientes del cáncer, Hanahan y Weinberg32 describieron seis características cardinales necesarias para la carcinogénesis, como la autosuficiencia en las seńales proliferativas, la insensibilidad a las seńales antiproliferativas, la evasión de las seńales apoptóticas normales, el automantenimiento de la angiogénesis, la invasión tisular y metástasis, y finalmente, el potencial ilimitado de duplicación. De esta manera, es la combinación de las mutaciones del ADN y la selección natural la que básicamente conduce a la evolución de un clon celular que ha adquirido estas seis características y que han sido exitosas para la transformación en una célula cancerosa.33,34
Hanahan y Weinberg sugirieron además que las células cancerosas podrían adquirir estas características esenciales a través de vías temporal y mecánicamente distintivas. Esto pareció sugerir un gran grupo de vías posibles por las cuales un colonocito podría alcanzar su transformación neoplásica. Esto difirió marcadamente de una hipótesis previa de Fearon y Vogelstein,35 quienes en 1990 sugirieron que los pasos hacia la carcinogénesis eran limitados; que ciertas mutaciones en los genes eran adquiridas con frecuencia y en un orden específico.
Desde entonces numerosos estudios han avalado y mejorado posteriormente el modelo de Vogelstein.2,30 A pesar de su amplia aceptación, este modelo no puede explicar las diferencias considerables que se reconoce que existen entre los cánceres. Quizás una síntesis de estos modelos puede probar ser el mejor abordaje (figura 1). En numerosos estudios se vio que la invasión del tejido es una característica tardía de la carcinogénesis del colon, mientras que el incremento de la proliferación y la reducción de la apoptosis se observan con frecuencia en forma temprana en los FCA.24-26,28,36-39
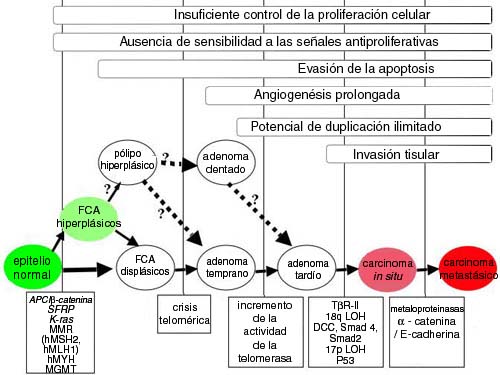
Figura 1.
Progresión escalonada hacia la carcinogénesis del colon humano. Según las características morfológicas, la progresión desde la mucosa colónica normal hasta el carcinoma aparece en forma de un patrón bastante predecible. Los focos crípticos aberrantes (FCA) displásicos son los precursores más tempranos; sin embargo, datos recientes sugieren que los pólipos y los FCA hiperplásicos podrían progresar hacia FCA displásicos y adenomas. Además, diversos fenómenos genéticos se pueden observar con frecuencia en pasos similares de la vía. La inestabilidad genética, con inclusión de los defectos en CIN, MIN y BER, y la mutagénesis epigenética se presentan, de manera característica, durante el estadio inicial de la progresión y proveen un entorno permisivo para que las células adquieran mutaciones adicionales, lo que provoca la adquisición de caracteres necesarios para la transformación carcinogénica. Durante esta progresión, diversas vías de regulación y de seńalización son blancos frecuentes en cualquiera de sus niveles, lo que puede alterar su expresión o función. Estas vías incluyen, entre otras, la Wnt/APC/β-catenina, BMP/TGF-β/SMAD, Src, y K-ras.
La angiogénesis y el potencial de duplicación ilimitado se hallan por lo general incrementados en los pólipos tempranos a tardíos.40-42 En resumen, la transformación neoplásica es un proceso escalonado en el cual las mutaciones del ADN y la selección clonal resultan en la evolución de una célula que expresa las características comunes a todas las células cancerosas.
Síndromes familiares de cáncer de colon
El estudio de los síndromes familiares de cáncer de colon y la identificación de las mutaciones genéticas transmisibles han sido de gran utilidad para nuestra comprensión del proceso de la carcinogénesis colónica. Con frecuencia se ha hallado que los genes identificados en los síndromes de cáncer familiares estaban mutados o silenciados en los cánceres colónicos esporádicos.30,43- 45 Mientras que el síndrome familiar de cáncer transmisible, la poliposis adenomatosa familiar (PAF) y el síndrome de cáncer de colon no polipoide (SCCNP) representan sólo el 5% de todos los cánceres de colon, la identificación de las mutaciones transmisibles predisponentes aportó luz sobre los genes cuya mutación es fundamental para la aparción de cánceres colónicos esporádicos.30,43 Más recientemente, se describió un nuevo síndrome de poliposis en los seres humanos, la poliposis adenomatosa MYH (PAM), y se identificaron las bases genéticas para diversos trastornos familiares asociados con cáncer colorrectal (tabla 1). Cuando se examinan minuciosamente, se puede notar en estos síndromes familiares el predominio de diversas vías de seńalización o regulatorias: Wnt/APC/β-catenina, TGF-β/BMP, falta de concordancia del ADN y mecanismos de reparación y escisión de bases. Todos los cánceres de colon familiares y esporádicos tendrán alteraciones significativas en al menos uno de estos mecanismos regulatorios y de seńalización.

Aunque estas vías son el blanco frecuente de fenómenos mutagénicos durante la carcinogénesis colorrectal no se las encuentra en todas las neoplasias. En consecuencia, deben existir otros mecanismos alternativos, todavía no reconocidos o no completamente comprendidos, para la progresión neoplásica del epitelio colónico.46-53 Los pacientes con enfermedad inflamatoria intestinal tienen riesgo de cáncer de colon a través de una vía alternativa, cáncer asociado a la colitis (CAC).54-57 Estas vías alternativas son importantes desde el punto de vista clínico y probablemente ampliarán nuestro conocimiento de la génesis tumoral. Sin embargo, nos concentraremos en el resto de los mecanismos de inestabilidad genética observados en la mayoría de los carcinomas colorrectales familiares y esporádicos.
Inestabilidad genética y epigenética
Aunque la acumulación de mutaciones somáticas del ADN es la fuerza de conducción detrás de la progresión al cáncer, la inestabilidad genética es con frecuencia la característica del medio que da lugar a estos eventos mutagénicos críticos.33,58-60 La inestabilidad genética ha sido observada en las etapas más tempranas de la tumorigénesis, los FCA.22,28,61 Ahora reconocemos que existen diversas formas distintivas de inestabilidad genética.58,59,62 En el presente, nuestra comprensión de estas formas es limitado; a medida que aprendemos más, será más probable que su importancia se incremente para los modelos de cáncer colorrectal.
La vasta mayoría de los tumores en los seres humanos son aneuploides. La aneuplodía está marcada por la pérdida o ganancia de cromosomas completos, un fenotipo descrito como inestabilidad cromosómica (INC).59 La pérdida de heterozigocidad (PDH) está asociada con este fenotipo; es la pérdida de un alelo de un gen. Así, solamente una copia de un gen necesita estar mutada somáticamente con la pérdida del alelo de tipo salvaje por la PDH. Las anormalidades cromosómicas de los tumores con INC también incluyen la amplificación génica y la translocación cromosómica. Si bien las causas de la INC son desconocidas, en los últimos ańos se han logrado avances significativos en el tema. Sin lugar a dudas, el dańo de los puntos de control del ADN desempeńa un papel en este proceso.60,63,64 Más aun, la proteína APC, además de su papel central en la PAF y en la carcinogénesis esporádica, está involucrada en la segregación cromosómica.65,66 El trabajo realizado más recientemente sobre la investigación del mantenimiento de los telómeros en células normales, así como de la supervivencia y multiplicación de las células neoplásicas aportó nuevas ideas hacia otro mecanismo potencial para explicar las inestabilidades observadas en los cánceres con INC.42 Los telómeros son complejos nucleoproteicos en los extremos de los cromosomas que funcionan para mantener la extremidad de éstos.42,67 También funcionan para limitar el número absoluto de divisiones celulares. La actividad de la telomerasa en los adultos está limitada a los linfocitos activados, células germinales y células germinales tisulares. En ausencia de actividad de la telomerasa, los telómeros se acortan progresivamente con cada división celular.
Después de un número limitado de divisiones, los telómeros quedan acortados de manera crítica y son inestables e inducen la apoptosis celular o la senescencia.42,68 Este período que provoca el acortamiento de los telómeros se denomina “crisis telomérica”. Si los puntos de control fallan, y si no son seguidos por senescencia o apoptosis, se observa inestabilidad cromosómica grave, con características similares a las observadas en las neoplasias humanas, como puentes cromosómicos, translocaciones no recíprocas, amplificación génica y rearreglos cromosómicos complejos.42,69 Los estudios de pólipos colónicos y de cánceres humanos hallaron que la actividad de la telomerasa es baja y que la longitud de los telómeros estaba acortada en pólipos de tamańo pequeńo a moderado. La actividad de la telomerasa está incrementada en los pólipos grandes y en los cánceres colorrectales,70,71 lo que permite un potencial duplicativo ilimitado, un “sello” del cáncer. Asociados con la baja actividad de la telomerasa, los estudios de hibridación genómica comparativa (HGC) y los histológicos informaron incremento en las tasas de rearreglos cromosómicos, translocaciones no recíprocas y puentes en la anafase observados en los pólipos adenomatosos.72 Por último, los estudios en animales transgénicos avalan esta hipótesis.
Los cánceres epiteliales espontáneos son infrecuentes en ratones salvajes y en ratones con mutación de p53, sin embargo en los ratones con esta mutación y deficientes en telomerasa existe un incremento notable de tumores epiteliales de mama, piel y tracto gastrointestinal. Además, estos cánceres se caracterizan por translocaciones no recíprocas y por otras anomalías citogenéticas asociadas con los tumores con INC.42,69,73,74 En conjunto, los estudios de tumores en seres humanos y los estudios en transgénicos sugieren que la crisis telomérica puede ser un mecanismo importante que provoca INC en cánceres con INC.
Quizá la mejor comprendida de las inestabilidades genéticas involucradas en la progresión del cáncer de colon sea el error en la determinación de la compatibilidad (RD).75-77 El sistema de RD reconoce las incompatibilidades base a base y los apareamientos desiguales de inserción/deleción que tienen lugar con la duplicación del ADN y con la recombinación homóloga. Estos errores deben ser corregidos antes de la duplicación del ADN, porque de otra manera las mutaciones pasarían a las células hijas. Segmentos cortos repetitivos de ADN, conocidos como microsatélites, son vulnerables a este tipo de error. En células con deficiencia por RD, con frecuencia estos sitios se encuentran mutados.58,59,76 Este tipo de inestabilidad genética se conoce como inestabilidad de microsatélites (INM o IMS). Los genes que contienen estas secuencias repetitivas en sus regiones de codificación son particularmente susceptibles a la inactivación de mutaciones durante la duplicación del ADN en neoplasias con IMS.78-81 Los cánceres con IMS, a diferencia de los tumores con INC, tienden a ser diploides o casi diploides.59,76 La INM puede aparecer tanto en cánceres de colon familiares como esporádicos, aunque los mecanismos responsables son bastante diferentes. El síndrome de Lynch (SCCNP) es un síndrome familiar de cáncer de colon con transmisión dominante75,82,83 y se caracteriza por la aparición del tumor a una edad temprana (media de 45 ańos) y tasas elevadas de neoplasias extraintestinales,75,82,84 como estómago, ovario, uréter, pelvis renal, cerebro (principalmente glioblastoma multiforme – también conocido como síndrome de Turcot–),85 intestino delgado, sistema hepatobiliar y piel (neoplasia de las glándulas sebáceas: síndrome de Muir- Torre).86 Se observa inactivación de mutaciones en líneas germinales en hMLH1 o hMLH2 en casi el 60% de las familias con SCCNP, y estas mutaciones de las líneas germinales también fueron descritas, aunque con una frecuencia menor, para hPMS1, hPMS2, hMSH6 y hMLH3.87-93 En contraste, en tumores esporádicos con INM, son infrecuentes las mutaciones somáticas en hMLH1, hMSH2 y hPMS2.94,95 Con mayor frecuencia, los niveles proteicos de hMLH1 están marcadamente disminuidos por un silenciamiento específico del gen por hipermetilación de los promotores.96,98 Tomadas en conjunto, las formas familiares y esporádicas de INM representan cerca del 15% al 20% de los cánceres de colon.59
Las bases del ADN se alteran continuamente por especies químicamente reactivas, desaminaciones hidrolíticas, metilaciones de guanina y alquilaciones. Esto puede llevar a la desigualdad en el apareamiento de los nucléotidos y a las mutaciones de transversión de bases (G → A o C → T) durante la duplicación del ADN si no se corrigen.99 La vía de la reparación de escición de bases (REB) es responsable de la identificación de estas bases con modificaciones químicas y de la iniciación de su reparación. Estas mutaciones de base única son con frecuencia silentes, sin embargo si tienen lugar en secuencias críticas de codificación pueden provocar mutaciones sin sentido. Las primeras pueden ser inactivadoras (por ejemplo en APC) o activadoras (por ejemplo, Ras).8,99,100 Las mutaciones sin sentido provocan acortamientos prematuros de la proteína si se encuentran dentro de la secuencia de codificación y son tipícamente inactivadoras. Diversos componentes de la vía de REB han sido recientemente involucrados en la patogénesis de la carcinogénesis colorrectal, como MED1 (MBD4), MGMT y hMYH.6-8,51,52,99,101-103
Mientras que las inestabilidades genéticas cromosómicas y microsatélites tienden a ser recíprocamente exclusivas, los defectos de escisión y reparación de bases pueden observarse en cánceres INC+ y INM+, así como en un pequeńo subgrupo de cánceres que son INC-/INM-.51,52,99 MED1(MBD4) es una glucosilasa involucrada en la reparación de las desaminaciones de las metilcitocinas. Se halla con frecuencia mutada por la INM en tumores INM+.102,104 Por el contrario, las mutaciones de la línea germinal hMYH están asociadas con un nuevo síndrome familiar atenuado de cáncer de colon que se asemeja a la PAF, ahora denominado PAM (poliposis adenomatosa MYH).6-8 Estos pacientes presentan, como característica, desde 15 a más de 100 pólipos, con una edad promedio de presentación a partir de los 40 ańos. Resulta sorprendente que la transmisión sea autosómica recesiva, con una penetrancia del cáncer de al menos el 50%.6,8 Los tumores de colon asociados con PAM son diferentes de los esporádicos y de los que están vinculados con PAF. Son diploides o casi diploides, y estables desde el punto de vista genético a nivel cromosómico y microsatelital. Además, se caracterizan por tasas frecuentes de mutaciones de transversión G → T, lo que provoca la inactivación de APC y la activación de K-ras.6-8,100 La inactivación de MYH también se asoció, recientemente, con la patogénesis del cáncer de colon esporádico.103 La metilguanina metil transferasa (MGMT) es otra enzima reparadora del ADN y un componente de la vía de REB.51,52 La MGMT elimina las lesiones alquilantes de O6-metilguanina, las cuales, si no se corrigen, pueden resultar en mutaciones de transición G → A. Las transiciones G → A son una causa frecuente de mutaciones K-ras.
En cánceres esporádicos, el gen MGMT está con frecuencia silenciado por la hipermetilación del promotor,51,52,99 en particular en aquellos con fenotipos INC- e INM-. En resumen, la reparación de las bases químicamente modificadas del ADN es una función fundamental que puede ser una fuente importante de inestabilidad genética en células en las cuales estas vías se hallan interrumpidas.
Por último, la regulación epigenética de la expresión génica mostró desempeńar un papel importante en la carcinogénesis y puede ser el episodio crítico que conduce a la progresión en un subgrupo de cánceres de colon esporádicos.105- 110 Los mecanismos epigenéticos incluyen la metilación del ADN, el sello (imprinting) del gen, la acetilación de las histonas, y son con frecuencia empleados para el silenciamiento de los genes y para la represión de la transcripción viral y de los trasposones. Los dinucleótidos CpG están presentes en los promotores de muchos genes y están destinados a la metilación por una clase de enzimas conocidas como metiltransferasas del ADN.108,109,111 Una vez que fueron metilados, una familia de proteínas que se conoce como MBDs (del inglés methyl-CpG binding domain proteins [proteínas ligadoras de metil-CpG]) se unen a los dinucléotidos CpG metilados y reclutan otros factores para formar un complejo que altera la conformación del ADN y de la cromatina hacia una configuración más estable y silente.
Mientras que la metilación CpG es en sí mutagénica (la desaminación de la metil- citosina puede provocar transiciones de los nucléotidos de C por T si no se corrigen), se cree que el silenciamiento de los genes supresores de los tumores y de la reparación del ADN mediante la hipermetilación del promotor puede ser el mecanismo predominante que promueve la carcinogénesis.106 En el cáncer de colon, p16lnk4a
P>(regula la actividad de ciclina D), MLH1 (un gen RD), p14ARFP>
(reguladora en más de la actividad de p53), APC, y O6-MGMT (que repara las mutaciones de guanina a adenosina), han sido identificadas como blancos comunes para el silenciamiento mediante la metilación de promotores.96,98,112-115 Ya que la mayoría de los cánceres de colon tendrán algún grado de hipermetilación de promotores, un subgrupo tendrá un grado elevado de esta hipermetilación en varios supresores tumorales simultáneamente. Se ha propuesto que este subgrupo constituye una vía separada para la tumorigénesis colorrectal, llamada isla CpG del fenotipo metilador (CIMP, por sus siglas en inglés
[CpG island methylator phenotype].105,116-118 Los tumores CIMP+ son típicamente diploides o casi diploides (INC-), y pueden ser INM+ (debido a la inactivación de hMLH1) o pueden tener la REB alterada debido a la reducción de los niveles de MGMT. Más aun, el fenotipo CIMP+ parece estar presente típicamente en adenomas dentados y en pólipos hiperplásicos grandes. La caracterización clínica y molecular de este fenotipo es un área de activas investigaciones.
Sinopsis
Desde la última vez que revisamos este tema se lograron grandes avances en relación con la comprensión de la carcinogénesis colorrectal. Quizá lo más interesante sea que mejoramos la idea que teníamos acerca de los mecanismos que gobiernan la inestabilidad genética y su papel en la progresión neoplásica.
Continuamos creyendo que esta progresión neoplásica tiene lugar en un número limitado de vías, en las cuales se hallan inactivados supresores tumorales específicos o los oncogenes están activados en una secuencia bastante definida.
Con la continuidad de las investigaciones veremos cambios sorprendentes en la forma de prevenir, diagnosticar y tratar esta enfermedad mortal.
Los autores no manifiestan “conflictos de interés”.
BIBLIOGRAFÍA
-
Parkin DM, Bray FI, Devesa SS. Cancer burden in the year 2000. The global
picture. Eur J Cancer 2001; 37 Suppl 8:S4-66.
-
Lynch JP, Hoops TC. The Genetic Pathogenesis of Colorectal Cancer.
Hematology - Oncology Clinics of North America 2002; 16(4):1-36.
-
Meyskens FL, Jr. Chemoprevention of FAP with sulindac. Curr Oncol Rep 2002;
4(6):463.
-
Phillips RK, Wallace MH, Lynch PM, Hawk E, Gordon GB, Saunders BP, et al. A
randomised, double blind, placebo controlled study of celecoxib, a selective
cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous
polyposis. Gut 2002; 50(6):857-60.
-
Asano TK, McLeod RS. Non steroidal anti-inflammatory drugs (NSAID) and
Aspirin for preventing colorectal adenomas and carcinomas. Cochrane Database
Syst Rev 2004(2):CD004079.
-
Sampson JR, Dolwani S, Jones S, Eccles D, Ellis A, Evans DG, et al. Autosomal
recessive colorectal adenomatous polyposis due to inherited mutations of MYH.
Lancet 2003; 362(9377):39-41.
-
Jones S, Emmerson P, Maynard J, Best JM, Jordan S, Williams GT, et al.
Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and
somatic G:C-->T:A mutations. Hum Mol Genet 2002; 11(23):2961-7.
-
Sieber OM, Lipton L, Crabtree M, Heinimann K, Fidalgo P, Phillips RK, et al.
Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line
mutations in MYH. N Engl J Med 2003; 348(9):791-9.
-
Halford SE, Rowan AJ, Lipton L, Sieber OM, Pack K, Thomas HJ, et al. Germline
mutations but not somatic changes at the MYH locus contribute to the pathogenesis
of unselected colorectal cancers. Am J Pathol 2003; 162(5):1545-8.
-
Pineau BC, Paskett ED, Chen GJ, Espeland MA, Phillips K, Han JP, et al. Virtual
colonoscopy using oral contrast compared with colonoscopy for the detection of
patients with colorectal polyps. Gastroenterology 2003; 125(2):304-10.
-
Mak T, Lalloo F, Evans DG, Hill J. Molecular stool screening for colorectal
cancer. Br J Surg 2004; 91(7):790-800.
-
Diaz-Rubio E. New chemotherapeutic advances in pancreatic, colorectal, and
gastric cancers. Oncologist 2004; 9(3):282-94.
-
Kim EC, Lance P. Colorectal polyps and their relationship to cancer.
Gastroenterol Clin North Am 1997; 26(1):1-17.
-
Carethers JM. The cellular and molecular pathogenesis of colorectal cancer.
Gastroenterol Clin North Am 1996; 25(4):737-54.
-
Winawer SJ, Zauber AG, Ho MN, O'Brien MJ, Gottlieb LS, Sternberg SS, et al.
Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp
Study Workgroup. N Engl J Med 1993; 329(27):1977-81.
-
Winawer SJ, Zauber AG, O'Brien MJ, Gottlieb LS, Sternberg SS, Stewart ET, et
al. The National Polyp Study. Design, methods, and characteristics of patients with
newly diagnosed polyps. The National Polyp Study Workgroup. Cancer 1992; 70(5
Suppl):1236-45.
-
Takayama T, Katsuki S, Takahashi Y, Ohi M, Nojiri S, Sakamaki S, et al.
Aberrant crypt foci of the colon as precursors of adenoma and cancer. N Engl J Med
1998; 339(18):1277-84.
-
Roncucci L, Pedroni M, Vaccina F, Benatti P, Marzona L, De Pol A. Aberrant
crypt foci in colorectal carcinogenesis. Cell and crypt dynamics. Cell Prolif 2000;
33(1):1-18.
-
Tudek B, Bird RP, Bruce WR. Foci of aberrant crypts in the colons of mice and
rats exposed to carcinogens associated with foods. Cancer Res 1989; 49(5):1236-
40.
-
Nucci MR, Robinson CR, Longo P, Campbell P, Hamilton SR. Phenotypic and
genotypic characteristics of aberrant crypt foci in human colorectal mucosa. Hum
Pathol 1997; 28(12):1396-407.
-
Siu IM, Pretlow TG, Amini SB, Pretlow TP. Identification of dysplasia in human
colonic aberrant crypt foci. Am J Pathol 1997; 150(5):1805-13.
-
Heinen CD, Shivapurkar N, Tang Z, Groden J, Alabaster O. Microsatellite
instability in aberrant crypt foci from human colons. Cancer Res 1996;
56(23):5339-41.
-
Pedroni M, Sala E, Scarselli A, Borghi F, Menigatti M, Benatti P, et al.
Microsatellite instability and mismatch-repair protein expression in hereditary and
sporadic colorectal carcinogenesis. Cancer Res 2001; 61(3):896-9.
-
Shivapurkar N, Huang L, Ruggeri B, Swalsky PA, Bakker A, Finkelstein S, et al.
K-ras and p53 mutations in aberrant crypt foci and colonic tumors from colon
cancer patients. Cancer Lett 1997; 115(1):39-46.
-
Shpitz B, Bomstein Y, Shalev M, Liverant S, Kaufman Z, Klein E, et al.
Oncoprotein coexpression in human aberrant crypt foci and minute polypoid lesions
of the large bowel. Anticancer Res 1999; 19(4B):3361-6.
-
Takayama T, Ohi M, Hayashi T, Miyanishi K, Nobuoka A, Nakajima T, et al.
Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma,
cancer, and familial adenomatous polyposis. Gastroenterology 2001; 121(3):599-
611.
-
Smith AJ, Stern HS, Penner M, Hay K, Mitri A, Bapat BV, et al. Somatic APC and
K-ras codon 12 mutations in aberrant crypt foci from human colons. Cancer Res
1994; 54(21):5527-30.
-
Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Dong Chen W, et
al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in
colorectal cancer. Nat Genet 2004; 36(4):417-22.
-
Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet 1993;
9(4):138-41.
-
Chung DC. The genetic basis of colorectal cancer: insights into critical pathways
of tumorigenesis. Gastroenterology 2000; 119(3):854-65.
-
Ponder BA. Cancer genetics. Nature 2001; 411(6835):336-41.
-
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100(1):57-70.
-
Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and
darwinian selection in tumours. Trends Cell Biol 1999; 9(12):M57-60.
-
Muto T, Bussey HJ, Morson BC. The evolution of cancer of the colon and
rectum. Cancer 1975; 36(6):2251-70.
-
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell
1990; 61(5):759-67.
-
Shiozawa J, Ito M, Nakayama T, Nakashima M, Kohno S, Sekine I. Expression
of matrix metalloproteinase-1 in human colorectal carcinoma. Mod Pathol 2000;
13(9):925-33.
-
Adachi Y, Yamamoto H, Itoh F, Hinoda Y, Okada Y, Imai K. Contribution of
matrilysin (MMP-7) to the metastatic pathway of human colorectal cancers. Gut
1999; 45(2):252-8.
-
Vermeulen SJ, Bruyneel EA, Bracke ME, De Bruyne GK, Vennekens KM,
Vleminckx KL, et al. Transition from the noninvasive to the invasive phenotype and
loss of alpha-catenin in human colon cancer cells. Cancer Res 1995; 55(20):4722-
8.
-
Portera CA, Jr., Berman RS, Ellis LM. Molecular determinants of colon cancer
metastasis. Surg Oncol 1998; 7(3-4):183-95.
-
Zhang X, Gaspard JP, Chung DC. Regulation of vascular endothelial growth
factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res 2001;
61(16):6050-4.
-
Kaklamanis L, Kakolyris S, Koukourakis M, Gatter KC, Harris AL. From
hyperplasia to neoplasia and invasion: angiogenesis in the colorectal adenoma-
carcinoma model. Adv Exp Med Biol 2000; 476:249-66.
-
Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J
Clin Invest 2004; 113(2):160-8.
-
Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996;
87(2):159-70.
-
Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and
caretakers. Nature 1997; 386(6627):761, 763.
-
Chung DC, Rustgi AK. DNA mismatch repair and cancer. Gastroenterology
1995; 109(5):1685-99.
-
Jass JR, Biden KG, Cummings MC, Simms LA, Walsh M, Schoch E, et al.
Characterisation of a subtype of colorectal cancer combining features of the
suppressor and mild mutator pathways. J Clin Pathol 1999; 52(6):455-60.
-
Hasegawa H, Ueda M, Watanabe M, Teramoto T, Mukai M, Kitajima M. K-ras
gene mutations in early colorectal cancer ... flat elevated vs polyp-forming cancer.
Oncogene 1995; 10(7):1413-6.
-
Olschwang S, Slezak P, Roze M, Jaramillo E, Nakano H, Koizumi K, et al.
Somatically acquired genetic alterations in flat colorectal neoplasias. Int J Cancer
1998; 77(3):366-9.
-
Saitoh Y, Waxman I, West AB, Popnikolov NK, Gatalica Z, Watari J, et al.
Prevalence and distinctive biologic features of flat colorectal adenomas in a North
American population. Gastroenterology 2001; 120(7):1657-65.
-
Yashiro M, Carethers JM, Laghi L, Saito K, Slezak P, Jaramillo E, et al. Genetic
pathways in the evolution of morphologically distinct colorectal neoplasms. Cancer
Res 2001; 61(6):2676-83.
-
Jass JR, Whitehall VL, Young J, Leggett BA. Emerging concepts in colorectal
neoplasia. Gastroenterology 2002; 123(3):862-76.
-
Jass JR. Hyperplastic polyps and colorectal cancer: is there a link? Clin
Gastroenterol Hepatol 2004; 2(1):1-8.
-
Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, Coxhead J, et al.
Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal
cancer. Proc Natl Acad Sci U S A 2002; 99(14):9433-8.
-
Hofseth LJ, Khan MA, Ambrose M, Nikolayeva O, Xu-Welliver M, Kartalou M, et
al. The adaptive imbalance in base excision-repair enzymes generates microsatellite
instability in chronic inflammation. J Clin Invest 2003; 112(12):1887-94.
-
Guo HH, Loeb LA. Tumbling down a different pathway to genetic instability. J
Clin Invest 2003; 112(12):1793-5.
-
Walsh S, Murphy M, Silverman M, Odze R, Antonioli D, Goldman H, et al. p27
expression in inflammatory bowel disease-associated neoplasia. Further evidence of
a unique molecular pathogenesis. Am J Pathol 1999; 155(5):1511-8.
-
Itzkowitz SH. Inflammatory bowel disease and cancer. Gastroenterol Clin North
Am 1997; 26(1):129-39.
-
Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev 2004;
23(1-2):11-27.
-
Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers.
Nature 1998; 396(6712):643-9.
-
Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers.
Nature 1997; 386(6625):623-7.
-
Augenlicht LH, Richards C, Corner G, Pretlow TP. Evidence for genomic
instability in human colonic aberrant crypt foci. Oncogene 1996; 12(8):1767-72.
-
Breivik J, Gaudernack G. Genomic instability, DNA methylation, and natural
selection in colorectal carcinogenesis. Semin Cancer Biol 1999; 9(4):245-54.
-
Murray AW. The genetics of cell cycle checkpoints. Curr Opin Genet Dev 1995;
5(1):5-11.
-
Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, et al.
Mutations of mitotic checkpoint genes in human cancers. Nature 1998;
392(6673):300-3.
-
Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, et al.
Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat
Cell Biol 2001; 3(4):433-8.
-
Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Nathke IS. A role for
the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol
2001; 3(4):429-32.
-
Chan SR, Blackburn EH. Telomeres and telomerase. Philos Trans R Soc Lond B
Biol Sci 2004; 359(1441):109-21.
-
Stewart SA, Weinberg RA. Telomerase and human tumorigenesis. Semin
Cancer Biol 2000; 10(6):399-406.
-
Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, et al. Telomere
dysfunction promotes non-reciprocal translocations and epithelial cancers in mice.
Nature 2000; 406(6796):641-5.
-
Engelhardt M, Drullinsky P, Guillem J, Moore MA. Telomerase and telomere
length in the development and progression of premalignant lesions to colorectal
cancer. Clin Cancer Res 1997; 3(11):1931-41.
-
Plentz RR, Wiemann SU, Flemming P, Meier PN, Kubicka S, Kreipe H, et al.
Telomere shortening of epithelial cells characterises the adenoma-carcinoma
transition of human colorectal cancer. Gut 2003; 52(9):1304-7.
-
Hermsen M, Postma C, Baak J, Weiss M, Rapallo A, Sciutto A, et al. Colorectal
adenoma to carcinoma progression follows multiple pathways of chromosomal
instability. Gastroenterology 2002; 123(4):1109-19.
-
Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and
evolution of intestinal carcinoma in mice and humans. Nat Genet 2001; 28(2):155-
9.
-
Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, et al. p53 deficiency
rescues the adverse effects of telomere loss and cooperates with telomere
dysfunction to accelerate carcinogenesis. Cell 1999; 97(4):527-38.
-
Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes.
Oncogene 2004; 23(38):6445-70.
-
Peltomaki P. Deficient DNA mismatch repair: a common etiologic factor for
colon cancer. Hum Mol Genet 2001; 10(7):735-40.
-
Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet
Dev 1999; 9(1):89-96.
-
Huang J, Papadopoulos N, McKinley AJ, Farrington SM, Curtis LJ, Wyllie AH, et
al. APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl
Acad Sci U S A 1996; 93(17):9049-54.
-
Miyaki M, Iijima T, Kimura J, Yasuno M, Mori T, Hayashi Y, et al. Frequent
mutation of beta-catenin and APC genes in primary colorectal tumors from patients
with hereditary nonpolyposis colorectal cancer. Cancer Res 1999; 59(18):4506-9.
-
Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, et al.
Microsatellite instability and mutations of the transforming growth factor beta type
II receptor gene in colorectal cancer. Cancer Res 1995; 55(23):5548-50.
-
Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al. Somatic
frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator
phenotype. Science 1997; 275(5302):967-9.
-
Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al.
Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch
syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96(4):261-8.
-
Lynch HT, Smyrk T, Lynch J. An update of HNPCC (Lynch syndrome). Cancer
Genet Cytogenet 1997; 93(1):84-99.
-
Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary
nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the
International Collaborative group on HNPCC. Gastroenterology 1999; 116(6):1453-
6.
-
Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, et al. The
molecular basis of Turcot's syndrome. N Engl J Med 1995; 332(13):839-47.
-
Kruse R, Rutten A, Lamberti C, Hosseiny-Malayeri HR, Wang Y, Ruelfs C, et al.
Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations
similar to that in hereditary nonpolyposis colorectal cancer families defined by the
Amsterdam criteria. Am J Hum Genet 1998; 63(1):63-70.
-
Liu B, Parsons R, Papadopoulos N, Nicolaides NC, Lynch HT, Watson P, et al.
Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer
patients. Nat Med 1996; 2(2):169-74.
-
Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, et
al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal
cancer. Nat Genet 1997; 17(3):271-2.
-
Akiyama Y, Sato H, Yamada T, Nagasaki H, Tsuchiya A, Abe R, et al. Germ-line
mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal
cancer kindred. Cancer Res 1997; 57(18):3920-3.
-
Huang J, Kuismanen SA, Liu T, Chadwick RB, Johnson CK, Stevens MW, et al.
MSH6 and MSH3 are rarely involved in genetic predisposition to nonpolypotic colon
cancer. Cancer Res 2001; 61(4):1619-23.
-
Kolodner RD, Tytell JD, Schmeits JL, Kane MF, Gupta RD, Weger J, et al. Germ-
line msh6 mutations in colorectal cancer families. Cancer Res 1999; 59(20):5068-
74.
-
Liu T, Yan H, Kuismanen S, Percesepe A, Bisgaard ML, Pedroni M, et al. The
Role of hPMS1 and hPMS2 in Predisposing to Colorectal Cancer. Cancer Res 2001;
61(21):7798-802.
-
Wu Y, Berends MJ, Sijmons RH, Mensink RG, Verlind E, Kooi KA, et al. A role
for MLH3 in hereditary nonpolyposis colorectal cancer. Nat Genet 2001; 29(2):137-
8.
-
Liu B, Nicolaides NC, Markowitz S, Willson JK, Parsons RE, Jen J, et al.
Mismatch repair gene defects in sporadic colorectal cancers with microsatellite
instability. Nat Genet 1995; 9(1):48-55.
-
Ma AH, Xia L, Littman SJ, Swinler S, Lader G, Polinkovsky A, et al. Somatic
mutation of hPMS2 as a possible cause of sporadic human colon cancer with
microsatellite instability. Oncogene 2000; 19(18):2249-56.
-
Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and
functional consequences of hMLH1 promoter hypermethylation in colorectal
carcinoma. Proc Natl Acad Sci U S A 1998; 95(12):6870-5.
-
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation
of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic
colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res
1997; 57(5):808-11.
-
Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, et al.
Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism
causing human MSI cancers. Proc Natl Acad Sci U S A 1998; 95(15):8698-702.
-
Jiricny J, Marra G. DNA repair defects in colon cancer. Curr Opin Genet Dev
2003; 13(1):61-9.
-
Lipton L, Halford SE, Johnson V, Novelli MR, Jones A, Cummings C, et al.
Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway.
Cancer Res 2003; 63(22):7595-9.
-
Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M, et
al. Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-
specific DNA N-glycosylase. J Biol Chem 2000; 275(42):32422-9.
-
Riccio A, Aaltonen LA, Godwin AK, Loukola A, Percesepe A, Salovaara R, et al.
The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with
microsatellite instability. Nat Genet 1999; 23(3):266-8.
-
Kambara T, Whitehall VL, Spring KJ, Barker MA, Arnold S, Wynter CV, et al.
Role of inherited defects of MYH in the development of sporadic colorectal cancer.
Genes Chromosomes Cancer 2004; 40(1):1-9.
-
Bader S, Walker M, Hendrich B, Bird A, Bird C, Hooper M, et al. Somatic
frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch
repair deficiency. Oncogene 1999; 18(56):8044-7.
-
Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis
Rev 2004; 23(1-2):29-39.
-
Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat
Rev Genet 2002; 3(6):415-28.
-
Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer.
Annu Rev Genomics Hum Genet 2002; 3:101-28.
-
Tycko B. Epigenetic gene silencing in cancer. J Clin Invest 2000; 105(4):401-7.
-
Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics
joins genetics. Trends Genet 2000; 16(4):168-74.
-
Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG.
Aberrant patterns of DNA methylation, chromatin formation and gene expression in
cancer. Hum Mol Genet 2001; 10(7):687-92.
-
Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene
2001; 20(24):3139-55.
-
Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of
human cancer. Cancer Res 2001; 61(8):3225-9.
-
Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA,
et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human
cancer. Cancer Res 2000; 60(16):4366-71.
-
Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins
DN, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA
methyltransferase by promoter hypermethylation is associated with G to A
mutations in K-ras in colorectal tumorigenesis. Cancer Res 2000; 60(9):2368-71.
-
Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, et al.
Aberrant CpG-island methylation has non-random and tumour-type-specific
patterns. Nat Genet 2000; 24(2):132-8.
-
Rashid A, Shen L, Morris JS, Issa JP, Hamilton SR. CpG island methylation in
colorectal adenomas. Am J Pathol 2001; 159(3):1129-35.
-
Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal
tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci U
S A 2000; 97(2):710-5.
-
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island
methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999;
96(15):8681-6.
-
Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet 2001;
10(7):721-33.
-
Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A
serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998;
391(6663):184-7.
-
Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, et al. Peutz-Jeghers
syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet
1998; 18(1):38-43.
-
Dong SM, Kim KM, Kim SY, Shin MS, Na EY, Lee SH, et al. Frequent somatic
mutations in serine/threonine kinase 11/Peutz-Jeghers syndrome gene in left-sided
colon cancer. Cancer Res 1998; 58(17):3787-90.
-
Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, et al.
Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998;
280(5366):1086-8.
-
Zhou XP, Woodford-Richens K, Lehtonen R, Kurose K, Aldred M, Hampel H, et
al. Germline mutations in BMPR1A/ALK3 cause a subset of cases of juvenile
polyposis syndrome and of Cowden and Bannayan-Riley-Ruvalcaba syndromes. Am
J Hum Genet 2001; 69(4):704-11.
|