Volumen 7, Número 3, Octubre 2004
![]()
![]()
|
|
|
|
|
|
||
|
Expertos Invitados |
|
|
|
|
 |
Columnista
Experto de SIIC Dr. Albert J. Czaja Professor of Medicine, Consultant in Gastroenterolgy & Hepatology. Hepatology, chronic hepatitis, autoimmune hepatitis. |
|
|
|
|
 |
Columnista
Experto de SIIC Dr. Xiao-Feng Sun Associate Prof. Pathology. |
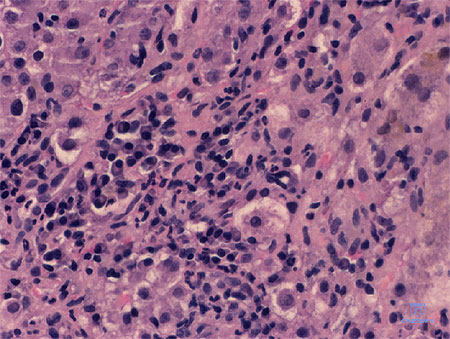
Introducción Figura 1. Expresión de p73 en línea celular de melanoma, por inmunotransferencia. La proteína p73 puede verse a los 73 kD.Inmunohistoquímica: Las secciones de bloques tisulares embebidos en parafina se desparafinaron con xileno y se rehidrataron. La actividad de la peroxidasa endógena se bloqueó con H2O2 al 0.5% en metanol. Con la finalidad de exponer los epitopes enmascarados, las secciones se incubaron con solución citrato (pH 6.0) a 80şC durante 10 minutos y luego se dejaron a temperatura ambiente. Luego de un lavado con PBS-Tween-20, los cortes se trataron con polvo bloqueante (BioGenex, San Ramon, CA, EE.UU.) durante 10 minutos para suprimir la fijación basal inespecífica. Luego del lavado, el primer anticuerpo diluido 1:150 en el diluyente (Dako Co, Glostrup, Dinamarca) se aplicó a 4şC durante toda la noche. Posteriormente, las secciones se incubaron con anti-IgG de cabra producida en asno y marcada con biotina durante 30 minutos, seguido de ABC de cabra (Santa Cruz Biotechnology) por otros 30 minutos. La reacción de peroxidasa se efectuó con el agregado de solución de tetrahidrocloruro de 3,3- diaminobencidina al 0.05% (Sigma Chemical Co, St. Louis, MO, EE.UU.) y H2O2 al 0.02% en PBS durante 8 minutos. Luego las secciones fueron teńidas con una segunda tinción de contraste. La mucosa normal análoga, los tumores primarios y las metástasis ganglionares se tińeron en la misma corrida de inmunofijación para evitar sesgo sobre el patrón e intensidad del teńido. En cada corrida con el primer anticuerpo o con PBS se incluyeron secciones que reaccionan con fuerza con el antip73, como controles positivos o negativos. En todos los procedimientos de fijación, los controles positivos mostraron una fijación manifiesta mientras que hubo ausencia completa de fijación en los controles negativos. Las muestras fueron analizadas en forma independiente por dos patólogos que desconocían datos clínicos e histológicos. Con la finalidad de evitar artefactos no se contaron las células en los márgenes de las secciones y las regiones con morfología pobre. Se consideró expresión negativa en el caso de ausencia de células tumorales positivas y equívoca cuando hubo menos de 10% de células tumorales positivas. Los casos positivos –con áreas de inmunorreactividad de más del 10%– se clasificaron como débiles, moderados y fuertes en virtud de la intensidad de fijación. Pérdida de heterocigotia: Se extrajo el ADN del tejido tumoral; el ADN de muestras correspondientes de mucosa normal fue heterocigota. Los productos de la reacción en cadena de polimerasa (PCR) se fragmentaron con Sty I (Fermentas, Vilna, Lituania), se controlaron en gel de agarosa al 3% y se compararon con el genotipo de mucosa normal. Análisis estadístico: Se usó la prueba de χ2 y el método de McNemar para determinar la significación de la diferencia en frecuencia de p73 entre muestras normales, tumor primario y metástasis, así como la asociación entre la expresión de p73 con otros factores clinicopatológicos. Se empleó el modelo de riesgo proporcional de Cox para estimar la relación entre la expresión de p73 y la supervivencia. Las curvas de descripción de supervivencia se computarizaron según el método Kaplan-Meier. Las pruebas fueron de dos colas y se consideró estadísticamente significativo un valor de p ? 5%. Resultados y discusión Analizamos la LOH en 52 tumores primarios y ninguno de ellos mostró LOH. Recientemente, Bengard y col. revisaron varios tipos de tumores, como cáncer colorrectal, gástrico, esofágico, hepatocelular, de mama, de cabeza y cuello, ovárico, renal, del sistema nervioso central, neoplasias hematológicas, melanoma y neuroblastoma y mostraron que la pérdida alélica (0.6% de 1 426 casos) y las mutaciones (20% de 1 049 casos) del gen p73 no son eventos genéticos principales en la carcinogénesis y el desarrollo tumoral. Todo indica que la p73 no funciona como gen supresor de tumores en la carcinogénesis. En función de las similitudes clinicopatológicas, en el estudio actual, los casos con tinción negativa o equívoca se consideraron negativos, mientras que los casos con fijación débil, moderada o fuerte se agruparon como positivos. La frecuencia de expresión de p73 aumentó desde mucosa normal (19%) a tumores primarios (67%) y a metástasis (95%, Figura 2). Aun en muestras pareadas, la frecuencia e intensidad de expresión de p73 estuvieron aumentadas desde tejido normal a tumor primario y metástasis (p ? 0.05, figura 3). Estudios previos mostraron que la transcripción de ARN mensajero de p73 está aumentada en tumores en comparación con el tejido adyacente normal, inclusove en cánceres colorrectales,4 mama,5 vejiga,6 pulmón7 y próstata.8 En conjunto, estos resultados indican que la p73 puede estar involucrada en la aparición y agresividad de los cánceres colorrectales. Sin embargo, la activación de alelos silenciosos o la expresión excesiva de p73, podría contribuir más en la carcinogénesis que con la supresión del tumor. 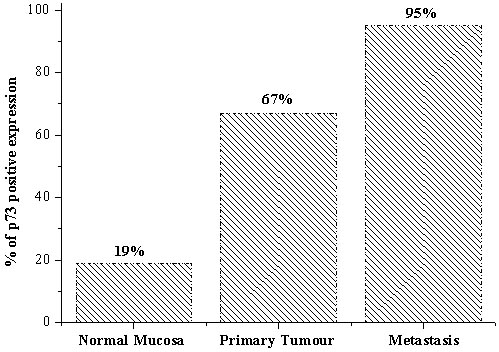 Figura 2. La frecuencia de expresión de p73 aumentó desde mucosa normal a tumor primario y a metástasis en ganglios linfáticos.  Figura 3. La mucosa normal no expresa la proteína p73 (a), las células del tumor primario correspondiente muestran fuerte expresión (b) y las metástasis correspondientes muestran fijación aun más marcada (c). Las secciones se tińeron también con hematoxilina.La asociación de la expresión de p73 con variables clinicopatológicas se presenta en la tabla 1. La expresión de p73 se correlacionó con la edad avanzada de los enfermos (p = 0.01). No pudimos encontrar correlación entre la expresión de p73, sexo, localización, estadio Dukes, patrón de crecimiento, diferenciación, infiltración y necrosis (p ? 0.05). Los pacientes con tumores p73 positivos tuvieron pronóstico significativamente más desfavorable respecto de los enfermos con tumores p73 negativos (p = 0.03, figura 4). En el análisis de variables múltiples, después del ajuste según parámetros clínicos y patológicos, el significado pronóstico de la expresión de p73 se mantuvo (p = 0.01, tabla 2). Liu y col. también encontraron que la mayor expresión de p73, determinada por inmunohistoquímica, se relacionó con sobrevida más corta en pacientes con cáncer colorrectal. Guan y col. revelaron que la expresión de p73 se relacionó con la angiogénesis en carcinomas colorrectales. No encontraron ninguna relación entre la expresión de p73, la diferenciación, el estadio, la localización y el tamańo.15,16 Tabla 1 Tabla 2 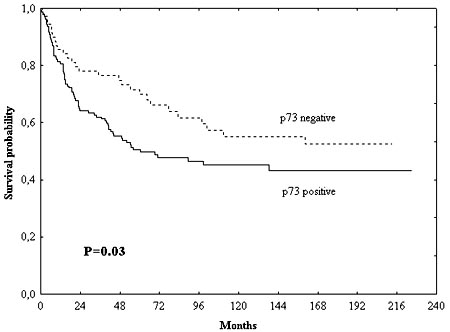 Figura 4. Los pacientes con tumores p73 positivos tuvieron supervivencia más corta que aquellos con tumores p73 negativos.En el estudio actual la expresión de p73 se correlacionó en forma positiva con la expresión de ras (p = 0.01). Más aun, en los pacientes con tumores ras positivos, la expresión de p73 predijo en forma positiva peor evolución que la negatividad de p73 (p = 0.02). No encontramos correlación entre la expresión de p73 y la expresión de p53, DCC, Bax, mutaciones de MBD4 y RIZ. Recientemente, Petrenko y colaboradores encontraron que la p73 coopera con el ras en la transformación primaria de fibroblastos in vitro y en la inducción de fibrosarcomas derivados de MEF in vivo en ratones sin timo. Aunque la p73 comparte su estructura y composición funcional con p53, existe una diferencia significativa en el desarrollo de tumores. En conclusión, la expresión de la proteína p73 es mayor durante la evolución de mucosa normal a tumores primarios y metástasis. Más aun, la expresión exagerada de p73 es un factor predictivo de pronóstico desfavorable en pacientes con cáncer colorrectal. Sin embargo, la LOH del gen no fue un factor importante en la aparición tumoral. El autor no manifiesta conflictos.
|
|
|
|
Trabajos Distinguidos, Gastroenterología , integra el Programa SIIC de Educación Médica Continuada |
![]()
Bienvenidos
a siicsalud
Acerca de SIIC Estructura de SIIC
Sociedad Iberoamericana de Información
Científica (SIIC)
Av. Belgrano 430, (C1092AAR), Buenos Aires, Argentina
atencionallector@siicsalud.com;
Tel: +54 11 4342 4901; Fax: +54 11 4331 3305.
Casilla de Correo 2568, (C1000WAZ) Correo Central, Buenos
Aires.
Copyright siicsalud© 1997- 2004, Sociedad Iberoamericana de Información Científica (SIIC)