Los anticonceptivos orales combinados (ACO) constituyen el grupo de fármacos más utilizado a nivel mundial; están compuestos por un agente estrogénico y un progestágeno. En Espańa se estima que entre 15% y 20% de las mujeres que siguen algún métod
o anticonceptivo optan por los ACO.1
La relación entre estos fármacos y la enfermedad tromboembólica venosa (ETEV) y arterial fue ya establecida en la década de los ’60.2,3 Desde entonces, numerosos estudios epidemiológicos demostraron claramente la asociación entre dichos tratamientos hormonales y el aumento del riesgo de trombosis, tanto en el territorio arterial4-7 como venoso.8-11 Algunos estudios han relacionado la dosis de estrógeno con este riesgo, que se estima en aproximadamente el doble en los ACO
que contenían más de 50 µg de etinilestradiol con respecto a aquellos con una cantidad inferior de estrógeno;10 no obstante, existen estudios de casos y controles y de cohortes que no arriban a la misma conclusión respecto de dicha relación
dosis-efecto.11,12
Durante las últimas décadas la investigación en este campo se centró en modificar la composición de los ACO con el objetivo de mejorar los efectos adversos metabólicos y reducir el riesgo de ETEV; de hecho, la reducción del componente estrogénico a 20-30
µg ha disminuido dicho riesgo.13 Sin embargo, los cambios introducidos en el tipo de progestágeno, que consisten en el empleo de agentes con menor acción androgénica (los llamados gestágenos de tercera generación), han compensado la reducción
del riesgo de ETEV conseguida con la disminución del contenido estrogénico e, incluso, lo han aumentado,14-16 lo cual reafirma la influencia de los progestágenos en el riesgo de ETEV y siembra de nuevo cierta inquietud con respecto al uso de
los nuevos ACO y su potencial trombogénico en una población joven. Dicho riesgo se ve notablemente incrementado por su interacción con determinados factores de riesgo trombótico, de los cuales uno de los más relevantes son los estados de trombofilia congénita.
Anticonceptivos orales y riesgo trombótico
Incidencia de ETEV
La incidencia basal de ETEV entre mujeres jóvenes que no emplean ACO oscila entre 0.4 a 0.8 por 10 000 mujeres/ańo.16-18 En mujeres que utilizan ACO con bajas dosis de estrógenos (< 35 µg) esta incidencia se eleva a 3 por 10 000 mujeres/ańo,19 lo cual supone que podemos esperar entre 20 y 40 casos anuales de ETEV no mortal por cada 100 000 mujeres que siguen dicho tratamiento. La mortalidad por ETEV en personas jóvenes hospitalizadas oscila entre 1% y 2%;20 en mujeres jóvenes en tratamiento con ACO esta cifra se estima más próxima al 1%.21 Por tanto, se puede concluir que si bien el riesgo absoluto de ETEV sigue siendo bajo (sobre todo si se valora en relación con la elevada eficacia anticonceptiva que lo
s ACO poseen) no debe ser ignorado, sobre todo en determinados grupos de mujeres con una mayor prediposición trombótica.
No existen datos suficientes sobre el riesgo de retrombosis en mujeres con antecedentes trombóticos, asociados o no al uso de ACO, debido a que la mayoría de los médicos tradicionalmente desaconsejan el uso de dichos fármacos en estos grupos de mujeres debido al temor a una recurrencia trombótica.22 En mujeres con antecedentes de ETEV que inician terapia hormonal sustitutiva sí se demostró incremento del riesgo de retrombosis con respecto a grupos control,23 si bien estos resultado
s son difícilmente extrapolables a mujeres que toman ACO.
Relación entre los distintos tipos de ACO y riesgo de ETEV
A pesar de que los ACO con bajas dosis de estrógenos suponen un descenso del riesgo trombótico con respecto a los que empleaban dosis altas, dicho riesgo sigue existiendo. Así, estudios que comparan mujeres que toman ACO con dosis bajas de estrógenos
con otras que no siguen ningún tipo de tratamiento hormonal concluyen que el riesgo relativo (RR) de sufrir TVP y TEP mortal es de 2.1;24 el de TVP no mortal, de 3.8,19 y el de tromboflebitis superficial, TVP y TEP en conjunto es d
e 2.7.13 No existen aún datos suficientes referidos al riesgo de ETEV inducido por el uso de ACO con dosis muy bajas de estrógenos (20 µg o incluso 15 µg), con el incoveniente adicional del inadecuado control del ciclo menstrual al que se asocia en numerosas mujeres.
La introducción de los gestágenos modificó de modo significativo el riesgo trombótico consecutivo a los ACO debido a sus efectos sobre la hemostasia y a la distinta capacidad que éstos tienen de contrarrestar los efectos protrombóticos de los estrógenos,
como posteriormente comentaremos. Los gestágenos de primera generación (noretisterona y linestrenol) se asociaban con dosis altas de estrógenos (≥ 50 µg). Los gestágenos de segunda (levonorgestrel) y de tercera generación (desogestrel, gestodeno, nor
gestimato) se asocian con dosis bajas de estrógenos (30 a 35 µg ).
Los ACO de segunda generación aumentan el riesgo tromboembólico alrededor de 4 veces con respecto al basal en mujeres que no emplean este tipo de fármacos. A principios de los ańos ’80 se introdujeron los ACO de tercera generación (que incluían desogestrel y gestodeno como progestágenos) en un intento por reducir el riesgo de complicaciones cardiovasculares y reducir los efectos adversos de tipo androgénico (aumento de peso, acné) y los cambios adversos en el metabolismo de las lipoproteínas. El empleo
de los gestágenos de tercera generación duplicó o triplicó el riesgo de ETEV con respecto a los de segunda generación (siempre asociados con bajas dosis de estrógenos) a pesar de las aparentemente mínimas modificaciones en su molécula, los resultados de
los distintos estudios son coincidentes en este extremo;16-18,25 además, se encontraron diferencias entre los distintos gestágenos de tercera generación, siendo el desogestrel el agente con mayor potencial trombogénico.16-18,25,26 La tabla I refleja las notables diferencias entre los riesgos absolutos de ETEV con distintos progestágenos entre sí, así como en relación con mujeres que no consumen ACO.

La influencia que el uso creciente de estos ACO de tercera generación pudiera tener sobre el incremento de la mortalidad por tromboembolismo venoso en mujeres jóvenes ha sido puesta de manifiesto en sendos inquietantes estudios realizados en Holanda y Reino Unido; este incremento se mostró más evidente en mujeres menores de 30 ańos.27,28 La Agencia Europea del Medicamento también advirtió acerca del aumento del riesgo de tromboembolismo venoso mortal y no mortal que este tipo de fármacos de u
so tan extendido puede entrańar.
Mención aparte merecen los combinados hormonales que incluyen el agente antiandrógeno acetato de ciproterona empleados para el tratamiento de acné, seborrea e hirsutismo leve; éstos han sido poco estudiados pero parecen inducir un riesgo de ETEV 4 veces
superior a los ACO de segunda generación,29 e igualmente superior incluso a los de tercera generación. En el estudio en marcha del grupo de Leiden (Multiple Environmental and Genetic Assessment of Genetic Risk Factors for Thrombosis [ME
GA]) el aumento del riesgo relativo de trombosis fue 18 veces superior al de las mujeres que no estaban expuestas a tratamientos hormonales.30
La tabla II resume los resultados de algunos de los principales estudios que evalúan el riesgo de ETEV asociado al uso de distintos ACO.

Variables que influyen en el riesgo trombótico
Como sabemos, toda mujer que toma ACO está expuesta a un riesgo de ETEV superior al riesgo basal descrito en mujeres de la misma edad que no consumen tratamientos hormonales; no obstante, dicho riesgo puede verse incrementado aun en mayor cuantía por
una serie de factores que a continuación revisaremos:
1. Duración del tratamiento con ACO
El riesgo tromboembólico es independiente de la duración del tratamiento10,31 y retorna al nivel basal a los tres meses de su suspensión.25
2. Exposición previa a tratamientos con ACO
El riesgo trombótico es mayor en mujeres que inician por primera vez un tratamiento anticonceptivo que en aquellas que ya han consumido algún tipo de ACO con anterioridad. Este riesgo es aun mayor en aquellas mujeres que inician un tratamiento con ACO co
n un preparado de tercera generación (RR 2.4 con respecto a un ACO de segunda generación).16,32
3. Tiempo transcurrido desde el inicio del tratamiento
Los distintos estudios coinciden en que el riesgo de ETEV es mayor durante las etapas iniciales del tratamiento; así dicho riesgo es 2 veces mayor durante el primer ańo, el período de máximo riesgo son los primeros 6 meses (RR 3).25,32,33 Este
efecto se ve multiplicado en el caso de los ACO de tercera generación y de las mujeres con defectos trombofílicos. Por las razones comentadas se sugirió que los ACO de segunda generación podrían considerarse de elección en mujeres que los consumen por primera vez,26 puede considerarse su sustitución en caso de conveniencia por un ACO de tercera generación una vez transcurrido el primer ańo de tratamiento. La tabla III resume los riesgos relativos de ETEV con los distintos tipos de ACO en virtud del tiempo transcurrido desde el inicio del tratamiento y el consumo previo de ACO.

4. Edad
Contrariamente a lo que ocurre con la trombosis en el territorio arterial, la edad no aparece en general como factor de riesgo para la ETEV en mujeres que consumen ACO.25 No obstante, existen estudios que encuentran mayor aumento del riesgo relativo de ETEV en los grupos de mujeres de menor edad que consumen ACO de tercera generación por primera vez (RR 7 veces mayor entre los 15 y los 19 ańos y 4 veces mayor entre los 20 y los 24 ańos con respecto a los ACO de segunda generación en mujeres d
e la misma edad).28
5. Obesidad
La obesidad constituye, como máximo, un factor de riesgo débil de ETEV.34
6. Indice de masa corporal
Un índice de masa corporal (IMC) elevado (> 25 kg/m2) supone un aumento del riesgo trombótico, sobre todo en mujeres que consumen ACO de tercera generación.25
7. Tabaco
No se demostró su influencia en el riesgo de ETEV en mujeres en tratamiento con ACO;25 este factor sí supone, sin embargo, un factor de riesgo importante para la trombosis en el territorio arterial.
8. Insuficiencia venosa periférica
Supone, como máximo, un factor de riesgo débil de ETEV.35
9. Embarazo previo
La hipótesis de que la exposición previa a niveles elevados de estrógenos durante el curso de uno o más embarazos podría suponer un aumento del riesgo trombótico asociado con el consumo de ACO con respecto a las mujeres que nunca han estado embarazadas n
o ha sido confirmada.18
10. Antecedente de hipertensión durante un embarazo previo
Algún estudio encontró mayor riesgo de ETEV en mujeres con esta complicación del embarazo (no en el caso de hipertensión arterial que ocurre fuera de este contexto).25
11. Historia familiar previa de ETEV
El RR ajustado para la edad, tanto en el caso de los ACO de segunda generación como en los de tercera, no mostró diferencias entre las mujeres con y sin historia familiar de ETEV (entendida como trombosis venosa en los padres o al menos uno de los herman
os) en el estudio de Bloemenkamp y col.18
12. Trombofilia
Existe un efecto sinérgico en lo que se refiere a riesgo trombótico entre los ACO y los defectos trombofílicos en general, si bien sobre este aspecto nos extenderemos más en profundidad posteriormente.
Patogenia de la ETEV asociada con anticonceptivos orales
No existe en la actualidad un mecanismo biológico que explique de modo convincente la patogenia de la ETEV secundaria a ACO. Se ha descrito una amplia gama de cambios en el sistema hemostático producidos por los distintos componentes hormonales de los AC
O, pero la correlación entre dichos cambios y la génesis del fenómeno trombótico no ha sido aún bien establecida.
La existencia de una relación entre la dosis de estrógeno y el riesgo trombótico ha sido hallada en algunos10,36 pero no en todos los estudios.11,12,25,37 En cualquier caso, a pesar de que el riesgo trombótico estimado de los ACO que contenían progestágenos de primera y segunda generación tendía a ser menor cuando se usaban combinados con estrógenos en dosis bajas en lugar de altas, el impacto global de los ACO con dosis bajas de estrógenos sobre el riesgo trombótico se vio afectado por el mayor riesgo asociado con los progestágenos de segunda generación. El perfil metabólico favorable de estos gestágenos con respecto a los de primera y segunda generación (aumento de la fracción HDL del colesterol y descenso de la LDL)38,39 convierte en inesperado el aumento del riesgo trombótico que inducen.
Tradicionalmente, el riesgo trombótico asociado a los ACO se relacionó con su componente estrogénico. Puesto que los preparados de segunda y tercera generación contienen la misma dosis baja de estrógenos, las diferencias en este aspecto entre unos y otro
s pueden reflejar un efecto específicamente derivado del componente progestágeno. Un reciente estudio holandés40 ha tratado de develar los efectos de los progestágenos solos sobre el sistema hemostático (principalmente sobre el importante sistema anticogulante natural de la proteína C, integrado por las proteínas C y S) y concluye que los progestágenos inducen cambios de naturaleza anticoagulante contrarios a los producidos por los preparados combinados (que inclinan el equilibrio hemostático
hacia el lado procoagulante). Los mencionados cambios de naturaleza anticoagulante son más acusados con los progestágenos de segunda generación (de mayor acción androgénica) que con los de tercera, por lo que se ha propuesto que lo que en realidad podría ocurrir es que los efectos procoagulantes del etinilestradiol son compensados en menor medida por los gestágenos de tercera generación (en especial el desogestrel) que por los de segunda, hecho en el que radica la diferencia en el riesgo trombótico der
ivado de unos y otros.
Globalmente se observa que los ACO producen tanto activación del sistema de la coagulación como del sistema fibrinolítico, demostrada por la elevación de marcadores moleculares, por lo que cabría esperar un equilibrio teórico entre ambos, el que no deber
ía suponer una tendencia protrombótica. No obstante, existe una amplia variabilidad individual en la magnitud de dichos cambios en uno u otro sentido, lo cual sugiere, por una parte, una influencia independiente de los ACO sobre ambos sistemas (hemostático y fibrinolítico) y, por otra, la ausencia de una regulación individual del equilibrio entre ambos expresada en la ausencia de correlación entre los cambios producidos en ambos sistemas. No parece existir, sin embargo, acción sobre la función plaquetaria.41
Los efectos de los ACO sobre el sistema hemostático parecen ser más pronunciados en mujeres que han sufrido con anterioridad algún episodio de ETEV asociado al uso de ACO que en mujeres previamente sanas; a aquéllas se las ha denominado “hiperrespondedor
as hemostáticas”.42
No existe gran experiencia con los anticonceptivos que contienen dosis de 15 a 20 µg de etinilestradiol, si bien parece que éstos ejercen similares efectos (aunque en menor cuantía) sobre el sistema de la hemostasia y que las mayores diferencias se encuentran a nivel del sistema fibrinolítico, potenciando su acción.43-45 No se demostró hasta el momento que estas pequeńas diferencias se traduzcan en un descenso del riesgo trombótico.
El mecanismo por el cual se producen los efectos de los estrógenos y gestágenos sobre los sistemas de la hemostasia y la fibrinólisis aún no ha sido develado; se supone que el mecanismo fundamental obedece a la alteración del aclaramiento hepático de las
proteínas involucradas en dichos sistemas.41,46 No se demostró ningún tipo de influencia hormonal sobre la expresión de los genes involucrados en la síntesis de elementos de la hemostasia (con excepción del factor XII)47 ni tampoco
sobre su expresión a nivel celular o tisular.41
Los cambios inducidos por los ACO sobre el sistema de la hemostasia y de la fibrinólisis pueden resumirse de la siguiente manera:
1. Sistema hemostático
En términos generales, el equilibrio resultante de los cambios inducidos por los ACO sobre este sistema produce una tendencia procoagulante (aumento de la génesis de trombina), debido a la elevación de determinadas proteínas procoagulantes (factores de l
a coagulación) y el descenso en los niveles plasmáticos de otras proteínas del sistema anticoagulante fisiológico (proteína S y antitrombina, fundamentalmente). La gran mayoría de estos cambios se producen dentro del rango de la normalidad de cada una de
las proteínas mencionadas, lo cual pone en entredicho su implicación teórica en el riesgo tromboembólico. De nuevo se debe insistir en que los gestágenos de tercera generación poseen un papel especialmente destacado en este aspecto.
Los principales cambios producidos en este sistema de la hemostasia son los siguientes: elevación de los niveles de los factores dependientes de la vitamina K inducidos por los gestágenos de tercera generación, en especial el factor VIIa,38,41
(los gestágenos de segunda generación carecen de acción en este nivel) pero también los factores II (protrombina), IX y X;48 elevación de los niveles de FVIII, factor de Von Willebrand y fibrinógeno;49 descenso de los niveles de proteína S total y libre, antitrombina e inhibidor de la vía del factor intrínseco (proteína que forma un complejo con el FVIIa inhibiendo la acción de la vía intrínseca). La acción sobre los niveles de proteína C no es relevante, pero existe una tendencia
a su elevación.49
Esta activación global del sistema hemostático se pone de manifiesto en la elevación de los niveles de determinados marcadores moleculares como los fragmentos 1+2 de la protrombina (F1+2) generados durante la activación de la protrombina, el fibrinopéptido A (producido durante la conversión del fibrinógeno en fibrina) o los complejos trombina-antitrombina. Al igual que en el caso del sistema fibrinolítico, no se conoce la relación entre los cambios experimentados en las proteínas involucradas en la hemostasia y los observados en los marcadores moleculares.
Todos los cambios mencionados se producen, aunque en menor cuantía, con los ACO con 20 µg de estrógenos, como ya mencionamos anteriormente.
2. Sistema fibrinolítico
La activación del sistema fibrinolítico inducida por ACO se interpreta como debida fundamentalmente a varios factores:38,43,49,50 descenso de los niveles de inhibidor del activador del plasminógeno-1 (PAI-1); descenso de los niveles de activador tisular del plasminógeno (t-PA:Ag); elevación de los niveles de plasminógeno.
Estos cambios se producen de modo más significativo con los ACO de tercera generación y, en el caso del PAI-1, su asociación con el desogestrel es más estrecha que con el gestodeno.49
A nivel de marcadores moleculares el estado hiperfibrinolítico se refleja en una elevación de los niveles plasmáticos del dímero-D y los productos de degradación del fibrinógeno, así como de los complejos plasmina-ALFA2 antiplasmina.38,45
,49,50
3. Resistencia a la proteína C activada
Durante los últimos ańos se ha dado a esta alteración trombofílica un papel central en la patogenia de la ETEV asociada con ACO. La resistencia a la proteína C activadda (RPCA) es un factor de riesgo trombótico descrito en 1993 en familias con trombosis
venosa inexplicada51 y debida en un 95% de los casos a una mutación en el gen del factor V de la coagulación (arginina por glutamina en la posición 506-FVR506Q-) conocida como factor V de Leiden.52
Los ACO producen, por un mecanismo no bien comprendido, RPCA adquirida, la cual supone un factor de riesgo trombótico. De nuevo cabe decir que esta alteración se produce en mayor magnitud con los ACO que contienen gestágenos de tercera generación en relación con los de segunda generación, lo cual se ha invocado como la principal justificación del mayor riesgo trombótico de los anticonceptivos más modernos. Con estos agentes los resultados de la prueba coagulativa de la RPCA ofrecen resultados similares
a los obtenidos en los portadores heterozigotas del factor V de Leiden.53
Por otra parte, existen datos para afirmar que el grado de RPCA producido por distintos preparados de segunda generación se correlaciona inversamente con la dosis de levonorgestrel, lo cual sugiere que las concentraciones elevadas de este gestágeno contr
arrestan el aumento de la RPCA.54
Anticonceptivos orales y trombofilia congénita
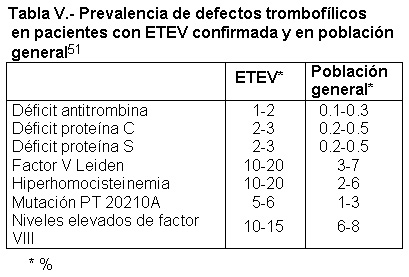
Durante las últimas décadas el conocimiento sobre los defectos trombofílicos se amplió notablemente (tabla IV), habiéndose investigado ampliamente el riesgo trombótico asociado con cada de uno de ellos, individualmente y formando combinaciones entre
sí, así como su interacción con determinados factores ambientales con los cuales actúan sinérgicamente aumentando el riesgo trombótico. La tabla V refleja la prevalencia de defectos trombofílicos en la población general y con ETEV. Los ACO son uno de los
factores de riesgo que han sido objeto de investigación más exhaustiva en lo que respecta a su riesgo trombótico per se y asociado con la existencia de determinados defectos trombofílicos.

En general, los ACO aumentan el riesgo de ETEV ocasionado por dichos defectos, el riesgo de dicha combinación es muy superior al riesgo individual inducido por cada uno de los factores por separado. Cada uno de los trastornos trombofílicos posee un perfil de riesgo trombótico diferente en su combinación con los tratamientos hormonales, lo cual debe ser tenido en cuenta de cara a la indicación del tratamiento anticonceptivo, por una parte, y al tipo de preparado, por otra. En términos generales debe contraindicarse el tratamiento con ACO en toda mujer que tenga algún defecto trombofílico, aun en los casos en los que éste no ha producido aún manifestaciones trombóticas; no obstante, esta afirmación admite algunos matices en algún caso concreto, como veremos. Igualmente es conveniente evitar el uso de ACO en mujeres con antecedentes personales (y posiblemente familiares) de trombosis. No existen estudios que aborden específicamente el riesgo trombótico de los nuevos preparados combinados hormonales de administración transdérmica, si bien probablemente no supongan una mejoría significativa en este aspecto.55
Los preparados exclusivamente sobre la base de gestágenos (desogestrel oral, implantes subdérmicos de etonogestrel o dispositivos de liberación intrauterina o intravaginal) podrían considerarse una opción válida en los mencionados grupos de pacientes si
se desea llevar a cabo una anticoncepción hormonal, teniendo en cuenta que con frecuencia producen incómodos cambios en los patrones de sangrado.56,57
La asociación entre ACO, trombofilia y trombosis venosa es también válida para la trombosis de los senos venosos cerebrales. La incidencia anual de este tipo de complicación en mujeres premenopaúsicas es muy baja (4 por 1 000 000). Los ACO elevan este riesgo hasta un odds ratio de entre 13 y 22, y de 30 si, además, se asocia algún defecto trombofílico con respecto a las mujeres que no presentan ninguno de estos dos factores de riesgo;58-60 por razones desconocidas este riesgo es máximo
para las portadoras de la mutación G20210A del gen de la protrombina (PT20210).60
Las mujeres con algún tipo de trastorno trombofílico que comienzan un tratamiento ACO desarrollan complicaciones trombóticas con mayor frecuencia y de modo más precoz tras el inicio; así, el riesgo relativo de ETEV con respecto al descrito para mujeres e
n tratamientos prolongados con ACO de tercera generación se incrementa 19 veces durante los 6 primeros meses de tratamiento y 11 veces durante el primer ańo. Por tanto, en ausencia de clínica trombótica previa la ETEV precoz en el curso de un tratamiento
con ACO obliga a descartar la presencia de un defecto trombofílico congénito.42 Esto da una idea de la importancia que el estudio de familiares sanos asintomáticos (sobre todo si son mujeres jóvenes) de pacientes con ETEV y trastornos trombofílicos tiene en la evaluación de este tipo de riesgos.
A continuación haremos breve referencia al riesgo trombótico de los ACO en mujeres con distintos trastornos trombofílicos:
Déficit de antitrombina
No existen series amplias de mujeres con este trastorno pero sí estudios limitados que demuestran con claridad que los ACO aumentan significativamente el riesgo de ETEV en mujeres con déficit de antitrombina (Tabla VI).61

Déficit de proteínas C y S
La escasa frecuencia de estos déficit hace que se hayan estudiado grupos pequeńos de pacientes sin que, quizá por esa razón, se haya demostrado un claro aumento del riesgo trombótico en estas mujeres (en especial aquellas con déficit de proteína S).61 No obstante, no puede excluirse un aumento del riesgo en estas mujeres, por lo cual se recomienda evitar tratamientos con ACO. La tabla VI muestra la incidencia de trombosis por paciente y ańo en mujeres con los déficit mencionados que sigue
n un tratamiento con ACO según el estudio de Pabinger y col.61
Resistencia a la proteína C activada (factor V de Leiden)
La relación entre el riesgo de ETEV y la ingesta de ACO en pacientes portadoras del factor V de Leiden (FVL) es la más ampliamente estudiada, debido fundamentalmente a la elevada prevalencia de dicha mutación en la población general, que ronda el 5%
en población de raza blanca del Reino Unido o el 2% en Espańa.62 Todos los análisis realizados demostraron la existencia una interacción sinérgica entre la presencia del FVL y el uso de ACO con respecto al riesgo de ETEV inducida por ambos factores de riesgo por separado, lo cual conduce a un RR multiplicado cuando ambos coinciden. La mutación heterozigota FVL se detecta en alrededor de un 30% de mujeres con ETEV que tienen lugar en el curso de tratamientos con ACO. El riesgo basal estimado d
e ETEV en mujeres no portadoras que no toman ACO es 0.8 por 10 000 mujeres/ańo; este riesgo aumenta hasta 3 por 10 000 mujeres/ańo entre quienes toman ACO pero no expresan FVL heterozigota (RR 3.7) y 5.7 por 10 000 mujeres/ańo en aquellas con FVL pero si
n ingesta de ACO (RR 6.9). En aquellas mujeres con ambos factores de riesgo la incidencia de ETEV se eleva a 28.5 por 10 000 mujeres/ańo (RR 34.7) con respecto a las mujeres no consumidoras ni portadoras del FVL; si hablamos de ACO que contienen desogestrel este RR llega hasta 50.18,19 Esto supone una mortalidad esperada en estos dos últimos grupos de mujeres de alrededor de 2.8 y 5 por 100 000 mujeres/ańo, respectivamente.19
La gran mayoría de los autores recomiendan emplear otras alternativas anticonceptivas en mujeres portadoras heterozigotas, si bien otros piensan que el riesgo trombótico es asumible siempre que no existan otros factores de riesgo para la trombosis, la paciente esté adecuadamente informada y no desee utilizar otros métodos de control de la natalidad.
En el caso de las portadoras homozigotas del FVL, el RR de ETEV cuando se asocia con ACO es de aproximadamente 100,63 por lo que la anticoncepción oral en este grupo de mujeres está totalmente contraindicada, sin excepciones.
La mutación G20210A del gen de la protrombina, cuya prevalencia en la población general se sitúa alrededor del 2%,64 es un factor de riesgo trombótico débil que se ve notablemente potenciado cuando se asocia a otros factores de riesgo trombótico, como los ACO. Las portadoras heterozigotas de esta mutación que consumen ACO presentan un aumento moderado del riesgo trombótico (RR 3.5) en relación con mujeres sanas que no toman ACO.65 Además, la combinación de ambos factores prod
uce de modo característico un aumento muy importante (del orden de 150 veces) del riesgo de trombosis venosa cerebral58 que, sorprendentemente, no se ve modificado por su asociación con el FVL;59 asimismo, se trata del único defecto
trombofílico para el que se ha descrito de modo característico un aumento en la incidencia de trombosis venosa de los miembros superiores vinculada con la ingesta de ACO.66
No existen datos con respecto a portadoras homozigotas y su relación con los ACO en cuanto al riesgo trombótico, pero es de esperar que el riesgo de esta combinación sea elevado. Las recomendaciones en cuanto a las precauciones de uso son similares a las
expresadas para el FVL heterozigota y homozigota.
Por otra parte, se ha descrito un aumento del riesgo de ETEV en mujeres consumidoras de ACO con niveles de factor VIII basales > 150 UI/dl no debidos a causa reactiva; además ambos factores poseen efectos aditivos, se encontró un odds ratio de 10.
3 (IC 95% 3.7-28.9) en relación con mujeres que no toman ACO y tienen niveles de factor VIII normales.67 Para valorar la modificación del riesgo trombótico que sufren las consumidoras de ACO con mutación homozigota C677T del gen la metilenetet
rahidrofolato reductasa no hay datos suficientes, si bien parece existir un efecto sinérgico entre ambos factores de riesgo trombótico, en particular en los casos en que la alteración genética se asocia con la presencia de hiperhomocisteinemia plasmática
.68,69 No se ha demostrado que los estrógenos influyan en los niveles de homocisteína plasmática.70 Tampoco existen datos basados en series amplias de pacientes con defectos trombofílicos combinados que evalúen el riesgo adicional d
e ETEV que suponen los ACO, si bien el elevado riesgo trombótico que entrańan contraindica dichos tratamientos en este grupo de mujeres.
Estudio de trombofilia previo al inicio de la anticoncepción oral
Las posibles complicaciones trombóticas potencialmente graves que pueden producirse en mujeres consumidoras de ACO con defectos trombofílicos han hecho surgir el interés por los estudios que evalúan la utilidad de la realización de pruebas de trombofilia congénita antes de la indicación de dicho tratamiento. Si esta medida se llevara a cabo su primera consecuencia sería la contraindicación del tratamiento ACO en una proporción no despreciable de mujeres (entre 3% y 6%), lo cual podría ocasionar embarazos no deseados debido a la falta de empleo de otros métodos anticonceptivos.22 Para la mayoría de las mujeres el riesgo trombótico asociado con los ACO es, a pesar de todo, bajo y aceptable, teniendo en cuenta los beneficios de un método anticonceptivo tan eficaz.
En general los análisis costo-beneficio de la realización sistemática de estudios de trombofilia previos a la anticoncepción oral no han resultado favorables a esta práctica, si bien la mayor discusión al respecto se plantea a propósito del FVL, como más
adelante comentaremos. Sin embargo, la historia trombótica personal y familiar (miembros de la familia que han sufrido episodios trombóticos recurrentes, a edad precoz o sin factores desencadenantes) es útil a la hora de iniciar un tratamiento de este tipo; el principal inconveniente de la historia familiar es su baja sensibilidad, sobre todo en el caso de familias pequeńas o de defectos trombofílicos que inducen riesgo trombótico moderado, como el FVL o la mutación PT20210 heterozigota.21,71
En el caso de los déficit de antitrombina, proteína C y proteína S, su baja prevalencia en la población hace que su determinación rutinaria con anterioridad a un tratamiento con ACO no sea recomendable en términos de costo-beneficio. El estudio de la mutación PT20210 tropieza con la dificultad de que, aun siendo ésta una anomalía prevalente en la población, no existe una prueba coagulativa que facilite y reduzca el costo de su diagnóstico, el cual en la actualidad sólo es posible por técnicas genéticas;
además el aumento del riesgo trombótico que induce en mujeres en tratamiento con ACO no pasa de ser moderado.
En virtud de su prevalencia en la población general, el efecto multiplicativo de su riesgo trombótico con los ACO y la existencia de una prueba coagulativa (RPCA) de fácil realización y excelente correlación con el diagnóstico genético (sensibilidad y es
pecificidad prácticamente del 100%)71 se ha planteado la determinación sistemática de este parámetro. Las conclusiones de la mayoría de los autores no justifican esta determinación en virtud de su costo-beneficio21,71,73,74 (se esti
ma que sería necesario estudiar alrededor de dos millones de mujeres para evitar una muerte por ETEV); en cambio, una minoría considera razonable la determinación sistemática de la RPCA y su confirmación por estudio genético de la mutación antes de iniciar un tratamiento con ACO, debido al costo personal y económico que la ETEV puede suponer en personas jóvenes y el riesgo de síndrome posflebítico tras un episodio de TVP en ellas (20% a 30% a los 5 ańos).1,75-77
BIBLIOGRAFÍA
-
Aznar J, Mira Y, Estellés A y col. Anticonceptivos orales y riesgo tromboembólico. Haematologica 2000; 85 (Supl. 2): 208-210.
-
Royal College of Practitioners. Oral contraceptives and thromboembolic disease. J Gen Coll Practit 1967; 13: 267-269.
-
Editorial. Oral contraceptives and thromboembolism. BMJ 1968; 2: 187-188.
-
Inman WH, Vessey MP, Westerholm B y col. Thromboembolic disease and the steroidal content of oral contraceptives: a report to the Committee on Safety of Drugs. 1970; 2: 203-209.
-
Jespersen J, Petersen KR, Skouby SO. Effects of newer oral contraceptives on the inhibition of coagulation and fibrinolysis in relation to dosage and type of steroid. Am J Obstet Gynecol 1990; 163: 396-403.
-
Stadel BV. Oral contraceptives and cardiovascular disease (first of two parts). N Engl J Med 1981; 305: 612-618.
-
Croft P, Hannaford PC. Risk factors for acute myocardial infarction in women: evidence from the Royal College of General Practitioners’ oral contraception study. BMJ 1989; 298; 165-168.
-
La Vecchia C, Franceschi S, Decarli Ay col. Risk factors for myocardial infarction in young women. Am J Epidemiol 1987; 125: 832-843.
-
Stolley PD, Strom BL, Sartwell PE. Oral contraceptives and vascular disease. Epidemiol Rev 1989; 11: 241-243.
-
Vessey MP, Doll R. Investigation of relation between use of oral contraceptives and thromboembolic disease. BMJ 1968; 2: 199-205.
-
Inman WHW, Vessey MP, Westerholm B y col. Thromboembolic disease and the steroidal content of oral contraceptives: a report to the Committee on Safety of Drugs. BMJ 1970; 2: 203-209.
-
Böttiger LE, Boman G, Eklund G y col. Oral contraceptives and thromboembolic disease: effects of lowering oestrogen content. Lancet 1980; 1: 1097-1101
-
Farmer RT, Preston TD. The risk of venous thromboembolism associated with low oestrogen oral contraceptives. J Obstet Gynaecol 1996; 15: 195-200
-
Spitzer WO, Lewis MA, Heinemann LAJ y col. Third generation oral contraceptives and risk of venous thromboembolic disease: An international case-control study. BMJ 1996; 312: 83-88.
-
Conard J. Biological coagulation findings in third-generation oral contraceptives. Hum Reprod Update 1999; 5: 672-680.
-
WHO Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception. Effect of different progestagens in low oestrogen oral contraceptives, on venous thromboembolic disease. Lancet 1995; 346: 1582-1588.
-
Jick H, Jick SS, Gurewich V y col. Risk if idiopathic cardiovascular death and non-fatal venous thromboembolism in women using oral contraceptives with differing progestagen components Lancet 1995; 36: 1589-1593.
-
Bloemenkamp KWM, Rosendaal FR, Helmerhorst FM y col. Enhacement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing third-generation progestagen. Lancet 1995; 346: 1593-1596.
-
Vandenbroucke JP, Koster T, Briët E y col. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet 1994; 344: 1453-1457.
-
Anderson FA, Wheeler B, Goldberg FJ y col. A population-based perspective of the hospital incidence and case-fatality rates of deep venous thrombosis and pulmonary embolism. Arch Intern Med 1991; 151: 933-938.
-
Vandenbroucke JP, van der Meer FJM, Helmerhorst FM y col. Factor V Leiden: should we screen oral contraceptive users and pregnant women? BMJ 1996; 313: 1127-1130.
-
Koster T, Small RA, Rosendaal FR y col. Oral contraceptives and venous thromboembolism: a quantitative discussion of the uncertainties. J Int Med 1995; 238: 31-37
-
Hoorbraten E, Qvistad E, Arnesen H y col. Increased risk of recurrent venous thromboembolism during hormone replacement therapy. Results of the randomized, double blind, placebo-controlled Estrogen in Venous Thromboembolism Trial. Thromb Haemost 2000
; 84: 956-961
-
Thorogood M, Mann J, Murphy M y col. Risk factors for fatal venous thromboembolism in young women: a case control study. Int J Epidemiol 1992; 21: 48-52.
-
World Health Organization Collaborative Study of Cardiovascular and Steroid Hormone Contraception. Venous thromboembolic disease and combined oral contraceptives: results of international multicentre case-control study. Lancet 1995; 346: 1575-1582.
-
Helmerhorst FM, Bloemenkamp KWM, Rosendaal FR y col. Oral contraceptives and thrombotic disease: risk of venous thromboembolism. Thromb Haemost 1997; 78: 327-333.
-
Vandenbroucke JP, Bloemenkamp KWM, Helmerhorst FM y col. Mortality from venous thromboembolism and myocardial infarction in young women in the Netherlands. Lancet 1996; 348: 401-402.
-
Thomas SHL. Mortality from venous thromboembolism and myocardial infarction in young adults in England and Wales. Lancet 1996; 348: 402
-
Vasilakis-Scaramozza C, Jick H. Risk of venous thromboembolism with ciproterone or levonorgestrel contraceptives. Lancet 2001; 358: 1427-1429.
-
Rosendaal FR, van Hylckama Vlieg A, Tanis BC y col. Estrogens, progestogens and trombosis. J Thromb Haemost 2003; 1: 1371-1380.
-
Vessey MP, Doll R. Investigation of relation between use of oral contraceptives and thromboembolic disease. A further report. BMJ 1969; 2: 651-657.
-
Poulter NR, Farly TMM, Chang CL y col. Safety of oral contraceptive pills. Lancet 1996; 347: 547
-
Bloemenkamp KWM, Rosendaal FR, Helmerhorst FM y col. Higher risk of venous thrombosis during early use of oral contraceptives in women with inherited clotting defects. Arch Intern Med 2000; 160: 49-52.
-
Stolley PD, Tonascia JA, Tockman MS y col. Thrombosis with low-estrogen oral contraceptives Am J Epidemiol 1975; 102: 197-208.
-
Royal College of Practitioners. Oral contraceptives, venous thrombosis and varicose veins. J Gen Coll Practit 1978; 28: 393-399.
-
Gerstman BB, Piper JM, Freiman JP y col. Oral contraceptive oestrogen and progestin potencies and the incidence of deep venous thromboembolism. Int J Epidemiol 1990; 4: 931-936.
-
Helmrich SP, Rosenberg L, Kaufman DW y col. Venous thromboembolism in relation to oral contraceptive use. Obstet Gynecol 1987; 69: 91-95.
-
Plu-Bureau G, Amiral J, Guize L y col. Safety of oral contraceptive pills. Lancet 1996; 347: 549.
-
Godsland IF, Crook D, Simpson R y col. The effect of different formulations of oral contraceptive agents on lipid and carbohydrate metabolism. N Engl J Med 1990; 323: 1375-1381
-
Kemmeren JM, Algra A, Meijers JC y col. Effect of second and third-generation oral contraceptives on the protein C system in the absence or presence of the factor V Leiden mutation: a randomized trial. Blood 2004; 103: 927-933
-
Kluft C, Lansink M. Effect of oral contraceptives on haemostasis variables. Thromb Haemost 1997; 78: 315-326.
-
Bloemenkamp KW, Rosendaal FR, Helmerhorst FM y col. Hemostatic effects of oral contraceptives in women who develop deep-vein thrombosis while using oral contraceptives. Thromb Haemost 1998; 80: 382-387.
-
Winkler UH, Schindler AE, Endrikat J y col. A comparative study of the effects on the hemostatic system of two monophasic gestodene oral contraceptives containing 20 µg and 30 µg ethinyl estradiol. Contraception 1996; 53: 75-84.
-
Basdevant A, Conard J, Pelissier C y col. Hemostatic and metabolic effects of lowering the ethinyl-estradiol dose from 30 µg to 20 µg in oral contraceptives containing desogestrel. Contraception 1993; 48: 193-204.
-
Winkler UH, Holscher T, Schulte H y col. Ethinylestradiol 20 versus 30 micrograms combined with 150 micrograms of desogestrel: a large comparative study of the effects of two low-dose oral contraceptives on the hemostatic system. Gynecol Endocrinol 1
996; 10: 265-271.
-
Kluft C. Fibrinolysis risk markers for cardiovascular disease. Gynecol Endocrinol 1993; 7 (Suppl.): 45-53.
-
Farsetti A, Misti S, Citarella F y col. Molecular basis of estrogen regulation of Hageman factor XII expression. Endocrinology 1995; 136: 5076-5083.
-
Daly L, Bonnar J. Comparative studies of 30 µg ethinylestradiol combined with gestodene and desogestrel on blood coagulation, fibrinolysis and platelets. Am J Obstet Gynecol 1990; 163: 430-437.
-
Gevers Leuven JA, Kluft C, Dersjant-Roorda MC y col. Effects of low dose oral contraceptives differing in progestogen compound on coagulation and fibrinolytic risk variables for venous and arterial thromboembolic diseases. En: Fibrinolysis and diseas
e, molecular and hemovascular aspects of fibrinolysis. Glas-Greenwalt P, ed CRC Press Chapter 32, 1995: 226-234.
-
Cachrimanidou AC, Hellberg D, Nilsson S y col. Hemostasis profile and lipid metabolism with long-interval desogestrel-containing oral contraceptive. Contraception 1994; 50: 153-165.
-
Dahlbäck B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: Prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA 1993;
90: 1004-1008.
-
Bertina RM, Koeleman BPC, Koster T y col. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369: 64-67.
-
Rosing J, Tans G. Effects of oral contraceptives on hemostasis and thrombosis. Am J Obstet Gynecol 1999; 180: S375-382
-
Kluft C, de Maat MP, Heinemann LA y col. Importance of levonorgestrel dose in oral contraceptives for effects on coagulation. Lancet 1999; 354: 832-833
-
Henzl MR, Loomba PK. Transdermal delivery of sex steroids for hormone replacement therapy and contraception. A review of principles and practice. J Reprod Med 2003; 48: 525-540.
-
Winkler UH, Howie H, Bühler K. A randomized controlled double-blind study of the effects on hemostasis of two progestogen-only pills containing 75 µg desogestrel or 30 µg levonorgestrel. Contraception 1998; 57: 385-392.
-
Egberg N. Effects on the hemostatic system and liver function in relation to Implanon and Norplant. Contraception 1998; 58: 93-98.
-
De Bruijn SF, Stam J, Koopman MM y col. Case-control study of risk of cerebral sinus thrombosis in oral contraceptive users who are carriers of hereditary prothrombotic conditions. BMJ 1998; 316: 589-592.
-
Martinelli I, Rosendaal FR, Vandenbrouck JP y col. Oral contraceptives are a risk factor for cerebral vein thrombosis. Thromb Haemost 1996; 76: 477-478.
-
Martinelli I, Sacchi E, Landi G y col. High risk of cerebral-vein thrombosis in carriers of the prothrombin-gene mutation and in users of oral contraception. N Engl J Med 1998; 338: 1793-1797.
-
Pabinger I, Schneider B and the GTH Study Group on Natural Inhibitors. Thrombotic risk of women with hereditary antithrombin-III, protein C and protein S deficiency taking oral contraceptive medication. Thromb Haemost 1994; 71: 548-552.
-
Rees D. The population genetics of factor V Leiden (Arg506Gln). Br J Haematol 1996; 95: 579-586.
-
Rosendaal FR, Koster T, Vandenbroucke JP y col. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995; 15: 1504-1508.
-
Zabalegui N, Montes R, Orbe J y col. Prevalence of FVR506Q and prothrombin 20210A mutations in the Navarrese population. Thromb Haemost 1998; 80: 522-523.
-
Aznar J, Vayá A, Estellés A y col. Risk of venous thrombosis in carriers of the prothrombin G20210A variant and factor V Leiden and their interaction with oral contraceptives. Haematologica 2000; 85: 1271-1276.
-
Vayá A, Mira Y, Mateo J y col. Prothrombin G20210A mutation and oral contraceptive use in upper-extremity deep vein trombosis. Thromb Haemost 2003; 89: 452-457.
-
Bloemenkamp KW, Helmerhorst FM, Rosendaal FR y col. Venous thrombosis, oral contraceptives and high factor VIII levels. Thromb Haemost 1999; 82: 1024-1027.
-
Chan HH, Douketis JD, Nowaczyk MJ. Acute renal vein thrombosis, oral contraceptive use, and hyperhomocysteinemia. Mayo Clin Proc 2001; 76: 212-214
-
Tonetti C, Ruivard M, Rieu V y col. Severe methylentetrahydrofolate reductase deficiency revealed by a pulmonary embolism in a young adult. Br J Haematol 2002; 119: 397-399.
-
Berger PB, Hermann RR, Dumesic DA. The effect of estrogen replacement therapy on total plasma homocysteine in healthy postmenopausal women. Mayo Clin Proc 200; 75: 18-23
-
Creinin MD, Lisman R, Strickler RC. Screening for factor V Leiden mutation before prescribing combination oral contraceptives. Fertil Steril 1999; 72: 646-651.
-
Tripodi A, Negri B, Bertina RM y col. Screening for the FV:Q506 mutation. Evaluation of thirteen plasma-based methods for their diagnostic efficacy in comparison with DNA-analysis. Thromb Haemost 1997; 77: 436-439.
-
Winkler UH, Zierleyin JP, Schulte H y col. Routine screening for coagulation inhibitors prior to prescribing the pill: prevalence data from a large cohort of German pill starters. Eur J Contracept Reprod Health Care 1996; 1: 47-52.
-
Rosendaal FR. Oral contraceptives and screening for factor V Leiden. Thromb Haemost 1996; 75: 524-525.
-
Aznar J, Gilabert J. Oral contraceptive use and screening of factor V Leiden. Thromb Haemost 1999; 81: 845-846.
-
Palareti G, Legnani C, Frascaro M y cols. Screening for activated protein C resistance before oral contraceptive treatment: a pilot study. Contraception 1999; 59: 293-299.
-
Prandoni P, Lensing AWA, Cogo A y cols. The long term clinical course of acute deep vein thrombosis. Ann Intern Med 1996; 125: 1-7.
|