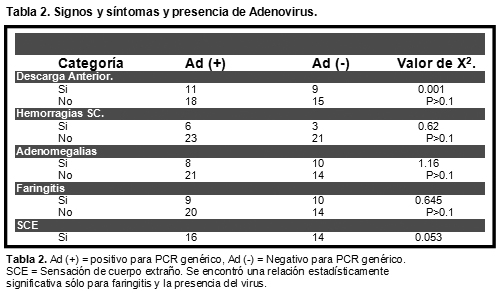
Introducción
Aproximadamente el 65% de todos los nińos con infección por VIH-1 o sida en los Estados Unidos son afroamericanos.1,2 Durante la era previa a la terapia antirretroviral de gran actividad (TARGA) más del 40% de los afroamericanos infectados por VIH experimentaba mayor cantidad de complicaciones renales que conducían a un lento crecimiento y a la progresión de la infección por VIH-1.3-15 Los pacientes infectados por VIH-1 corren riesgo de presentar insuficiencia renal aguda, glomerulopatías asociadas con trastornos inmunes, microangiopatías trombóticas asociadas al VIH y un síndrome renal denominado nefropatía asociada al VIH (NAVIH).3-7 Esta nefropatía fue descrita inicialmente en 1984 en pacientes infectados por VIH provenientes de Nueva York y Miami4,5 y se convirtió en una causa frecuente de enfermedad renal terminal (ERT) en los afroamericanos jóvenes.6-14 Esta nefropatía produce un impacto clínico adverso importante sobre la calidad de vida de los pacientes infectados por VIH. Lamentablemente, a pesar de los nuevos tratamientos antirretrovirales eficaces, seguimos viendo nińos con diagnóstico reciente de NAVIH en los Estados Unidos. Por todas estas razones es necesario conocer los mecanismos básicos responsables de la patogenia de la NAVIH y prevenir esta enfermedad. En esta revisión, analizaremos las cuestiones patogénicas más pertinentes y los aspectos controversiales relacionados con la patogenia de la nefropatía infantil asociada al VIH.
Definición
Esta nefropatía se caracteriza clínicamente por la presencia de proteinuria importante, síndrome nefrótico y rápida progresión hasta la insuficiencia renal crónica y se observa típicamente en pacientes afroamericanos.6,7 Al inicio, estos pacientes no muestran edema ni hipertensión importante a pesar de la proteinuria considerable y de la enfermedad glomerular. No obstante, se puede observar a menudo edema e hipertensión durante los estadios tardíos de la enfermedad renal. La histología renal muestra glomérulos con esclerosis focal y segmentaria, hipertrofia e hiperplasia de las células epiteliales viscerales y lesiones tubulointersticiales caracterizadas por túbulos microquísticos llenos de material proteináceo y células mononucleares infiltrantes.9-13 Estos cambios tubulares microquísticos conducen a agrandamiento renal, hallazgo que contrasta con los pequeńos rińones fibróticos observados en los casos típicos de pacientes con nefropatía crónica de otra etiología. Otro rasgo considerado característico de la NAVIH es la presencia de una glomerulopatía colapsante focal asociada con inclusiones tubulorreticulares en las células endoteliales y pérdida de diferenciación o proliferación de las células epiteliales glomerulares.9-11 Sin embargo, los nińos pueden mostrar sólo hiperplasia mesangial combinada con la dilatación tubular microquística al inicio de la proteinuria nefrótica.11-14 Más aun, al parecer, la enfermedad renal en los nińos con pruebas histológicas de hiperplasia mesangial podría progresar a ritmo más lento cuando es comparada con aquella que muestra pruebas iniciales de glomeruloesclerosis focal y segmentaria (GEFS).12,13 No se determinó aún si la hiperplasia mesangial es el primer paso hacia la GEFS o, como alternativa, puede evolucionar como un proceso patogénico independiente asociado con la infección viral.
El virus VIH-1
Algunos artículos anteriores de revisión analizaron con gran detalle varios conceptos fundamentales relacionados con la estructura del virus VIH-1.1,2 Aquí sólo revisaremos algunas ideas importantes relacionadas con el genoma del VIH-1 que son necesarias para comprender los conceptos analizados en esta revisión. Se considera el virus VIH-1 un retrovirus porque lleva ARN y, como tal, es necesario que invierta el flujo de la información genética en las células infectadas para generar ADN, transcritos de ARN viral y proteínas virales. El genoma del VIH-1 contiene por lo menos nueve genes que producen proteínas virales.1,2 Estas proteínas están divididas en proteínas estructurales, reguladoras y accesorias. Tres proteínas estructurales forman la microcápside viral, producen enzimas involucradas en la transcripción del VIH o producen glucoproteínas de superficie que participan en la unión a la célula y la entrada en ella. Estas proteínas estructurales se denominan Gag, Pol y Env, respectivamente.1,2 Dos proteínas reguladoras denominadas Tat y Rev, son esenciales para la infección por VIH, su replicación, y para la regulación del corte y empalme de los transcritos de ARN del VIH.1,2 Por último, cuatro proteínas accesorias denominadas Nef, Vif, Vpu y Vpr, desempeńan papeles esenciales en la replicación del VIH y la infección de las células.1,2 La patogenia de la enfermedad renal ha sido relacionada desde el punto de vista patogénico por lo menos con cuatro de estas proteínas, Env, Tat, Nef y Vpr.16-25 Explicaremos luego con más detalle su papel en la patogenia de la NAVIH.
Cambios proliferativos en el epitelio renal inducidos por el VIH-1
Como se explicó antes, un rasgo característico de la NAVIH es la presencia de rińones agrandados con microquistes renales y proliferación de las células epiteliales de los túbulos y los glomérulos.4,5,10-15 Sin embargo, los mecanismos patogénicos responsables de estas lesiones no se conocen de forma completa y han sido tema de muchos estudios y controversias. Una de las principales hipótesis actuales para explicar el modo en que el VIH-1 induce proliferación de las células epiteliales renales propone que VIH-1 puede inducir una infección productiva de los podocitos y las células epiteliales renales,26-29 y que la expresión de nef o vpr en estas células conduce a la estimulación directa de vías de seńalización del huésped que llevan a la proliferación celular.20-22 Sin embargo, esta hipótesis se basa en estudios in vitro realizados en el podocito murino inmortalizado condicionalmente y no está claro cómo se pueden aplicar estos hallazgos a la situación humana.20-22 Algunos estudios más recientes han sugerido que las células epiteliales parietales glomerulares son células en proliferación en el paciente que presenta una glomerulopatía colapsante asociada al VIH.15 Además, quedan varios interrogantes sin respuesta. Se desconoce el modo en que el VIH-1 puede inducir una infección productiva de las células epiteliales renales humanas in vivo. Algunos investigadores no pudieron detectar transcritos de ARN de HIV-1 en células epiteliales renales provenientes de pacientes con NAVIH.30-33 Tampoco está claro de qué modo los bajos niveles y el patrón focal de la infección por VIH-1 detectados en cortes renales de la NAVIH26-29 pueden explicar los cambios epiteliales proliferativos difusos observados en estos pacientes.
Proliferación de células epiteliales renales en ratones transgénicos para VIH (VIH-Tg)
La observación clave inicial que sugirió que los genes del VIH-1 eran responsables de inducir cambios proliferativos directos en células epiteliales renales se generó en el modelo VIH-Tg26.34 Estos ratones poseen una estructura defectuosa de ADN proviral de VIH-1, d1443, que carece de una secuencia de 3 kb que superpone las secuencias gag y pol,35,36 por lo tanto el VIH-1 no puede replicarse en estos ratones. Los ratones VIH-Tg26 nacen con rińones normales y muestran transcritos de ARN de VIH-1 en las células epiteliales glomerulares y tubulares.34-36 Las ratas VIH-Tg que tienen una estructura similar del ARN de VIH-1 también presentan cambios renales similares.37-38 Sobre la base de estos hallazgos, se utilizaron estos modelos transgénicos para explorar los mecanismos básicos por medio de los cuales el VIH-1 induce la proliferación de células epiteliales renales e identificar los productos virales involucrados en este proceso.19-22,34-39 Sin embargo, se obtuvieron diferentes resultados según se utilizaran células epiteliales renales primarias o inmortalizadas provenientes de estos modelos.20-22,35-40
Nosotros observamos que el primer estadio de la enfermedad renal se caracteriza por la presencia de lesión del epitelio tubular renal correlacionado con los altos niveles de expresión de los genes del VIH-1 en estas células.34,38 Además, nuestros hallazgos en células primarias apoyan la hipótesis de que los cambios proliferativos del epitelio renal constituyen un acontecimiento tardío en la patogenia de la enfermedad renal.34,38 Estos cambios proliferativos tardíos parecen ser impulsados, al menos en parte, por el aumento de los proteoglucanos de heparán sulfato renales y la acumulación de factores de crecimiento que fijan heparina.34,38 En concordancia con la idea de que el VIH-1 induce primero lesión renal, Brugemman y col. comunicaron que la activación de la expresión de los genes del VIH-1 mediante irradiación ultravioleta inducía apoptosis en células del epitelio tubular de VIH-Tg26.39 En conjunto, estos estudios sugieren firmemente que la activación directa de genes de HVI-1 en el epitelio tubular primario de VIH-Tg26 disminuye su velocidad de crecimiento o induce apoptosis.
Por oposición a estos hallazgos, los podocitos y las células epiteliales renales inmortalizadas condicionalmente tomadas de ratones VIH-Tg26 endogámicos con ratones immortomouse, que portan el antígeno T SV-40 termosensible [ts-SV-40-T] inducible por interferón, tuvieron cambios proliferativos notables siempre que se activaban genes VIH-1.20-22 Además, estos estudios identificaron al gen nef como causa de los cambios proliferativos.20-22 Sin embargo, incluso bajo estas condiciones, los efectos promotores del crecimiento del gen nef requieren cofactores adicionales presentes en el suero o la matriz extracelular de células confluentes. Globalmente, quedan varias preguntas por responder para validar la hipótesis de que el gen nef induce cambios proliferativos directos en las células epiteliales renales humanas. Por ejemplo, no está claro si las células epiteliales renales humanas expresan niveles importantes de la proteína Nef y si las células epiteliales renales primarias humanas responden al gen nef de forma similar a las células epiteliales murinas inmortalizadas condicionalmente.
Finalmente, algunos estudios recientes en ratones transgénicos sugirieron que la expresión de los genes del VIH-1 en los podocitos aislados puede conducir al espectro completo de la NAVIH.25 Sin embargo, estos estudios deben ser interpretados con precaución dado que estos ratones transgénicos expresan niveles muy altos de genes del VIH-1, y no se sabe si tales niveles de expresión del gen del VIH representan verdaderamente la situación clínica de los pacientes infectados por VIH. Por último, la presencia de microquistes tubulares en nińos infectados por VIH sin evidencia histológica de lesión de los podocitos sugiere firmemente que las células epiteliales del túbulo renal podrían ser afectadas de modo independiente de los cambios en los podocitos.
Papel de los factores renales locales en la patogenia de la NAVIH
Se cita a menudo un estudio crítico de Bruggeman y col. como prueba firme de que los factores renales locales por sí solos son suficientes para la presentación de la NAVIH.39 Este estudio muestra que los rińones de ratones VIH-Tg26 trasplantados a ratones silvestres evolucionan a NAVIH, mientras que los rińones de ratones silvestres trasplantados a ratones VIH-Tg26 no presentan la nefropatía.39 Sin embargo, debemos seńalar que este estudio no descarta la posibilidad de que algunos factores circulantes puedan desempeńar un papel en la patogenia de la NAVIH. En todos los modelos de VIH-Tg, se pasa por alto el proceso infeccioso inicial y la respuesta inmune del huésped y, por lo tanto, las células renales intrínsecas no sufren el impacto de la respuesta inmune sistémica. Además, las células endoteliales del ratón parecen no ser permisivas para la expresión de transcritos de ARN de VIH-1,40,41 y los ratones VIH-Tg26 no presentan inclusiones tuborreticulares en las células endoteliales renales, lesión característica observada en los seres humanos con NAVIH que se sabe es producida por la respuesta endotelial inmunitaria del huésped al virus. Más aun, los ratones VIH-Tg26 no liberan proteínas virales en la circulación y sus células mononucleares no producen las proteínas Env o Tat del VIH.34-36
En conjunto, estos hallazgos sugieren que el endotelio de los ratones VIH-Tg26 no es afectado por la presencia de transcritos virales, proteínas virales circulantes ni la respuesta inmune del huésped al VIH-1. Esta situación contrasta mucho con la situación clínica observada en los nińos infectados por VIH, que frecuentemente presentan una lesión endotelial progresiva secundaria a la respuesta inmune del huésped, infecciones oportunistas o el reclutamiento renal de células infectadas por VIH y proteínas virales circulantes.42-46 Por lo tanto, los rińones de ratones silvestres normales trasplantados en animales VIH-Tg26 no están expuestos a los efectos tóxicos potenciales de los productos virales circulantes, o las células mononucleares activadas o infectadas. Además, las células epiteliales renales de animales VIH-Tg26 pueden sufrir las consecuencias directas de la expresión de los genes del VIH-1 aun en ausencia de otros factores que son esenciales para permitir la infección o la lesión de las células epiteliales humanas.
Por último, vale la pena mencionar que no todos los ratones que expresan altos niveles de genes del VIH-1 en las células epiteliales renales presentan nefropatía. Gharavi y col. elaboraron híbridos F1 de VIH-Tg26FVB/N con otras cinco cepas de ratones endogámicos y observaron una notable variación del fenotipo renal de los ratones VIH-Tg26.47 Estos hallazgos demuestran una fuerte influencia genética que modula el resultado del fenotipo de la NAVIH en los ratones transgénicos y apoyan la idea de que además de los genes del VIH-1, otros factores genéticos o ambientales todavía desconocidos son esenciales para el desarrollo del fenotipo completo de la NAVIH en los seres humanos.48 Existe poco desacuerdo en la comunidad científica en relación con el concepto de que los antecedentes genéticos desempeńan un papel clave en la patogenia de la NAVIH. En resumen, muchos datos experimentales sugieren firmemente que los factores renales locales por sí solos no son suficientes para inducir el fenotipo completo de la NAVIH.
Papel de las proteínas virales circulantes y los factores de crecimiento fijadores de heparina
La activación y la disfunción de las células mononucleares o endoteliales infectadas por el VIH son eventos patogénicos importantes en la progresión del sida.42-46 Los nińos infectados por VIH a menudo desarrollan una lesión endotelial progresiva y crónica secundaria a la respuesta inmunitaria del huésped y a la presencia de infecciones oportunistas. Estos cambios aumentan la activación y la adhesión de las células mononucleares a las células endoteliales. Ambos tipos celulares pueden liberar múltiples citocinas y proteínas virales en la circulación sistémica o el rińón y estos factores circulantes pueden inducir una lesión renal por sí solos. Por ejemplo, Shirai y col. mostraron que los ratones VIH-Tg26 inmunizados con gp160 purificada recombinante mostraban una disminución del edema, la proteinuria y las concentraciones de BUN en suero.16 Estos autores sugirieron que su esquema de inmunización protegía a estos ratones contra los efectos adversos renales potenciales de gp160;16,17 sin embargo, hasta lo que sabemos, esta interesante hipótesis no ha sido probada y sus hallazgos con la inmunización no han sido reproducidos por otros grupos.
Como alternativa, los niveles circulantes del factor de crecimiento de los fibroblastos (FGF-2) básico están aumentados en los pacientes infectados por VIH-1 y se correlacionan con la progresión del sida45 y el desarrollo de la NAVIH.46 El FGF-2 es miembro de la familia de los factores de crecimiento de los fibroblastos que fijan heparina y ha sido implicado en la patogenia del sarcoma de Kaposi del sida.49,50 Algunos estudios anteriores mostraron que el FGF-2 participa en el desarrollo renal, la angiogénesis, la regeneración tubular y la progresión de varias nefropatías en seres humanos y roedores.50-62 El FGF-2 es producido por casi todos los tipos de células renales intrínsecas50,59-62 y por los linfocitos T y los macrófagos.53 El FGF-2 carece de un péptido seńal para la secreción, pero puede ser liberado por mecanismos no convencionales,49,50 que incluyen lesión celular, apoptosis, citocinas y proteasas liberadas por células infectadas por VIH o activadas. Una vez que el FGF-2 es liberado en el espacio extracelular, su actividad biológica es disminuida por su estrecha unión a los proteoglucanos de heparán sulfato.50 De esta forma, el FGF-2 se puede acumular potencialmente en el tejido renal y puede ser protegido de la degradación proteolítica. Tanto las células endoteliales lesionadas como las células mononucleares infectadas por VIH pueden liberar FGF-2 en la circulación de los pacientes infectados por VIH.45,46
En 1994, nosotros informamos una acumulación importante de FGF-2 en el túbulo-intersticio de ratones VIH-Tg26 con enfermedad renal en estadio tardío.34 Estos cambios se asociaron con un aumento de los proteoglucanos de heparán sulfato y el desarrollo de notables lesiones proliferativas del epitelio tubular renal.34 Estos hallazgos proporcionaron la primera prueba clara de que los rińones infectados por VIH podían actuar como una pileta que atrapaba factores de crecimiento fijadores de heparina liberados en la circulación o por células que infiltraban el rińón. Así, los proteoglucanos de heparán sulfato pueden actuar como un reservorio bioactivo que potencia la acumulación y la actividad de los factores de crecimiento fijadores de heparina en el rińón; cortes renales provenientes de nińos con NAVIH muestran un aumento considerable de los proteoglucanos de heparán sulfato renales y acumulación de FGF-2.46 Más recientemente, Tang y col. mostraron que el FGF-2 aumenta el reclutamiento renal y la fijación de las células mononucleares infectadas por VIH a las células epiteliales del túbulo renal in vivo,62 y que el FGF-2 aumenta la unión de las células mononucleares infectadas por VIH a células epiteliales del túbulo renal primarias cultivadas obtenidas de nińos con infección por VIH y nefropatía.62
En conjunto, estos estudios sugieren que el FGF-2 puede tener un nuevo papel inmunomodulador y de infectividad renal en la patogenia de la nefropatía infantil asociada al VIH. En apoyo de este concepto, Conaldi y col. mostraron que la proteína Tat del VIH aumenta la proliferación de los podocitos humanos cultivados al inducir la liberación de FGF-2 de su superficie celular.18 Estos hallazgos apoyan firmemente la hipótesis de que los proteoglucanos de heparán sulfato, VIH-Tat y FGF-2 desempeńan papeles sinérgicos en la patogenia de la nefropatía infantil asociada al VIH. Otro mecanismo que puede liberar tanto FGF-2 como VIH-Tat de sus sitios de almacenamiento de proteoglucanos de heparán sulfato es la digestión de los glucosaminoglucanos por proteasas y heparanasas.50,60,61 Estas enzimas son liberadas por las células mononucleares de sangre periférica infectadas por VIH o activadas, pero se desconoce su papel en la patogenia de la NAVIH.
Más recientemente, describimos que FGF-2 puede ser liberado por su unión a una proteína fijadora del factor de crecimiento de los fibroblastos (FGF-BP-1) que está aumentada en las células epiteliales del túbulo renal de nińos con NAVIH.63-64 En conjunto, al menos cuatro mecanismos diferentes pueden explicar la acumulación de FGF-2 en el rińón de nińos infectados por VIH con nefropatía: 1) la liberación de FGF-2 en la circulación por las células endoteliales lesionadas o las células inflamatorias activadas, 2) la unión del factor de crecimiento que fija heparina a los proteoglucanos de heparán sulfato renales, 3) la liberación de FGF-2 de las membranas basales y las células del túbulo renal en regeneración, 4) el aumento de FGF-BP-1 en las células epiteliales tubulares en proliferación y las células mononucleares que infiltran el rińón.
Papel de los proteoglucanos de heparán sulfato en la infección por VIH
En 1998 proporcionamos la primera prueba de que se podía provocar una infección productiva in vitro de las células epiteliales del túbulo renal humano obtenidas de nińos infectados por VIH cuando eran expuestas a aislados de VIH-1 primarios obtenidos de nińos con NAVIH.65 Sin embargo, es necesario seńalar que se requirió una carga viral elevada para infectar estas células y no pudimos identificar un correceptor importante del VIH-1 que participara en este proceso. Conaldi y col. confirmaron nuestros hallazgos en células epiteliales del túbulo renal obtenidas de pacientes VIH-negativos, pero atribuyeron el proceso infeccioso a la presencia de altos niveles de proteína CD4 sobre la superficie de estas células.66 En los estudios de seguimiento, ni otros autores ni nosotros pudimos detectar niveles importantes de receptores CD4 sobre las células epiteliales del túbulo renal y, por lo tanto, no quedan claros los mecanismos por medio de los cuales el VIH-1 entra en las células epiteliales del túbulo renal humano.67-71 Es tentador especular que los proteoglucanos de heparán sulfato pueden facilitar el ingreso del VIH-172-74 a las células epiteliales renales a través de vías endocíticas, como se demostró en células endoteliales cultivadas.77 Los proteoglucanos de heparán sulfato también pueden aumentar la infectividad del VIH-1 al facilitar la unión de este virus a la superficie celular.74-84 La afinidad de la proteína de la envoltura del VIH-1 gp120 por la heparina depende de la conservación de su bucle V3 y está influida firmemente por la carga del bucle V3.76 Por lo tanto, el VIH-1 puede utilizar los proteoglucanos de heparán sulfato como receptores de unión de baja afinidad para rastrear en la superficie celular receptores específicos para la entrada.79,80 En apoyo de este concepto, algunos polianiones solubles como heparina, sulfato de dextrán y polisulfato de pentosán pueden neutralizar la infección por VIH-1 in vitro.75,76 La infección por virus X4 parece ser más sensible a la inhibición por sulfato de dextrán que la infección por virus R5.73-76 Como alternativa, otras quimiocinas involucradas en la patogenia del sida (p. ej., SDF-1α, RANTES y M1F1-β) también pueden unirse a proteoglucanos de heparán sulfato y modular el proceso de infección por VIH.71,78-85 El VIH-Tat también puede imitar la acción de estas quimiocinas, puede unirse a la heparina con una afinidad similar a FGF-283-84 y requiere proteoglucanos de heparán sulfato para la internalización.85 Globalmente, es tentador especular que los proteoglucanos renales pueden actuar como una pileta que atrapa quimiocinas y VIH-1 de la circulación y de esta forma podría acelerar la progresión de la NAVIH.
Papel del VIH-Tat y los proteoglucanos de heparán sulfato en la NAVIH
El producto genético Tat es un factor de transcripción,86 pero también puede ser liberado en la circulación por células infectadas y captado por células no infectadas.87 El Tat liberado en el espacio extracelular (ecTat) puede inducir la proliferación de muchos tipos celulares.88-90 Como seńalamos antes, los proteoglucanos de heparán sulfato pueden actuar como receptores para la captación de ecTat. Este evento podría conducir a la activación del VIH-1 en las células mononucleares.71 Se sabe que todas las actividades biológicas de Tat in vitro, como inducción de crecimiento celular, migración e invasión, son aumentadas por las bajas concentraciones de heparina e inhibidas por sus altas concentraciones, como se demostró para FGF-2 y otros factores de crecimiento que fijan heparina.88-90 De forma similar, las citocinas liberadas por las células infectadas por VIH (es decir, interferón γ, factor de necrosis tumoral α e interleuquina 2) pueden inducir la síntesis o la liberación de FGF-2 y VEGF-A, un poderoso factor de crecimiento angiógeno y de permeabilidad.90-92 Estos factores actúan sinérgicamente con Tat, probablemente a través de la activación de las integrinas αvβ3 y α5β1 y las metaloproteinasas (MMP) MMP-2 y MMP-9.90-93 Una vez activadas, las MMP pueden desplazar el FGF-2 ligado a los proteoglucanos de heparán sulfato e inducir la proliferación de células.92 Debemos seńalar que los podocitos, al contrario de las células epiteliales del túbulo renal, tienen una capacidad in vivo limitada para sufrir división nuclear y citocinesis en respuesta al FGF-2.93-94
Muchas de estas células pueden mantenerse en la fase G1 tardía del ciclo celular.94 Por lo tanto, es tentador especular que cuando los podocitos diferenciados son inducidos a volver a entrar en el ciclo celular bajo la influencia de VIH-Tat o FGF-2, podrían sufrir cambios en el citoesqueleto que conducen a su desprendimiento, desdiferenciación, proliferación o muerte celular.94 De esta forma, VIH-ecTat puede alterar aun más la capacidad de los podocitos para responder a FGF-2. Los podocitos de afroamericanos infectados por VIH o de pacientes con predisposición genética a la nefropatía48 pueden ser más sensibles a los efectos de FGF-2 y VIH-Tat. Los estudios futuros deben identificar los factores genéticos y ambientales que podrían aumentar potencialmente los efectos adversos de las proteínas del VIH y los factores de crecimiento que fijan heparina en los pacientes infectados por VIH.
Pronóstico y futuro
En la era previa a la TARGA, los nińos con NAVIH progresaban muy rápidamente hasta la enfermedad renal terminal o morían en menos de 2 ańos.12,13 Actualmente, la institución temprana de TARGA conduce a la reducción de la carga viral y mejora el pronóstico de todas las enfermedades marcadoras del sida y prolonga la vida de los nińos infectados por VIH.14 Aproximadamente el 5% de los nińos afroamericanos infectados por VIH tratados con TARGA en nuestra institución, localizada en Washington DC, muestran pruebas clínicas o histológicas de NAVIH.14 La TARGA parece ser un tratamiento promisorio para prevenir el desarrollo o disminuir la progresión de la nefropatía.95,96 Debido a la TARGA algunos de estos nińos pueden tolerar las pérdidas de proteínas en la orina sin presentar edema ni enfermedad renal terminal.14 Además, el uso de inhibidores de la enzima convertidora de angiotensina (ECA) puede proporcionar beneficios terapéuticos adicionales.97 Por lo tanto, si los adelantos terapéuticos conducen a una mejoría importante en las tasas de supervivencia, la prevalencia de la NAVIH en los pacientes infectados puede aumentar siempre que estos nińos se tornen resistentes a la TARGA o no adhieran al tratamiento. El aumento de la prevalencia de pacientes infectados por VIH que se encuentran en la etapa previa a la enfermedad renal terminal o en ella en los Estados Unidos podría convertirse en un problema importante de salud pública. Estos pacientes correrán un riesgo elevado de morir por infecciones relacionadas con el VIH y complicaciones cardiovasculares y se desconocen las consecuencias cardiovasculares a largo plazo del uso de TARGA en los nińos.
Por último, los resultados promisorios comunicados en los receptores de trasplante renal infectados por VIH son alentadores.98,99 Sin embargo, hasta ahora, se sometieron a trasplante principalmente pacientes con otros tipos de nefropatías asociadas al VIH y no está claro si los pacientes con NAVIH responderán de forma similar. Además, sólo se consideran para el trasplante renal pacientes infectados por VIH muy seleccionados.98,99 Estos pacientes mostraban una adhesión excelente a la TARGA, ausencia de inmunosupresión grave (CD4 > 200 células/μl), viremia no detectable (< 50 copias de ARN de VIH-1/ml), ausencia de enfermedad marcadora de sida, tuvieron una reconstitución inmune exitosa después de la TARGA y no presentaron infecciones virales activas por lo menos durante los seis meses previos al trasplante renal. Lamentablemente, estos pacientes no son representativos de la población de infectados por VIH que se someten a diálisis en los Estados Unidos. Más aun, en la población pediátrica será difícil encontrar pacientes con NAVIH que reúnan estos criterios estrictos para el trasplante renal. Por lo tanto, la rápida identificación y el tratamiento inmediato de las mujeres embarazadas infectadas por VIH debe seguir siendo una prioridad absoluta para prevenir la transmisión infantil del VIH-1 y la nefropatía infantil asociada al VIH.
BIBLIOGRAFÍA
1. Fauci SA. HIV and AIDS. 20 years of science. Nature Med 9:839-843, 2003.
2. Gallo RC. Human retrovirus after 20 years; a perspective from the past and prospects for their future control. Immunol Rev 185:236-265, 2002.
3. Rao TKS, Friedman EA, Nicastri AD. The types of renal disease in the acquired immunodeficiency syndrome. N Engl J Med 16:1062-1068, 1987.
4. Rao TK, Filippone EJ, Nicastri AD, Landesman SH, Frank S, Chen CK, Friedman EA. Associated focal and segmental glomerulosclerosis in the acquired immunodeficiency syndrome. N Engl J Med 310:669-673, 1984.
5. Pardo V, Aldana M, Colton RM, Fischl MA, Jaffe D, Moskowitz L, et al. Glomerular lesions in the acquired immunodeficiency syndrome. Ann Intern Med 101:429-434, 1984.
6. Pardo V, Meneses R, Ossa L, Jaffe DJ, Strauss J, Roth D, et al. AIDS-related glomerulopathy. Occurrence in specific risk groups. Kidney Int 31:1167-1173, 1987.
7. Kopp JB, Winkler C. HIV-associated nephropathy in African Americans. Kidney Int 83:S43-S49, 2003.
8. Freedman BI, Soucie JM, Stone SM, Pegram S. Familial clustering of end stage renal disease in blacks with HIV-associated nephropathy. Am J Kidney Dis 34:254-258, 1999.
9. Barisoni L, Kriz W, Mundel P, D'Agati V. The dysregulated podocyte phenotype: A novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerolusclerosis and HIV-associated nephropathy. J Am Soc Nephrol 10:51-61, 1999.
10. Bourgoignie JJ, Pardo V. The nephropathology in HIV-1 virus infection. Kidney Int 40(35):S19-S23, 1991.
11. D'Agati V, Appel GB. HIV infection and the kidney. J Am Soc Nephrol 8:138:152, 1997.
12. Strauss J, Abitol C, Zilleruelo G, Scott G, Paredes A, Malaga S, et al. Renal disease in children with acquired immunodeficiency syndrome. N Engl J Med 321:625-630, 1989.
13. Ray PE, Rakusan T, Loechelt BJ, Selby DM, Liu XH, Chandra RS. HIV-1 associated nephropathy in children from the Washington, D.C. area: 12 years' experience. Semin Nephrol 18:396-405, 1998.
14. Ray PE, Xu L, Rakusan T, Liu XH. A 20-year history of childhood HIV-associated nephropathy. Pediatr Nephrol 19:1075-1092, 2004.
15. Dijkman HB, Weening JJ, Smeets B, Verrijp KC, Van Kuppevelt TH, Assmann KK, et al. Proliferating cells in HIV and pamidronate-associated collapsing focal segmental glomerulosclerosis are parietal epithelial cells. Kidney Int 70:388-344, 2006.
16. Shirai A, Klinman DM. Immunization with recombinant gp160 prolongs the survival of HIV-transgenic mice. AIDS Res and Hum Retroviruses 9:979-983, 1993.
17. Singhal PC, Sagar S, Chandra D, Garg P. Human immunodeficiency virus-1 gp120 and gp160 envelope proteins modulate mesangial cell gelatinolytic activity. Am J Pathol 147:25-32, 1995.
18. Conaldi PG, Botelli A, Baj A, Serra C, Fiore L, Federico G, et al. Human Immunodeficiency virus-1 Tat induces hyperproliferation and dysregulation of renal glomerular epithelial cells. Am J Pathol 161:53-61, 2002.
19. Hanna Z, Kay DG, Rebai N, Guimond A, Jothy S, Jolicoeur P. Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell 95:163-175, 1998.
20. Husain M, Gussella GL, Klotman ME, Gelman IH, Ross MD, Schwartz EJ, et al. HIV-1 Nef induces proliferation and anchorage-independent growth in podocytes. J Am Soc Nephrol 13:1806-1815, 2002.
21. Sunamoto M, Husain M, He JC, Schwartx EJ, Klotman PE. Critical role for Nef in HIV-1 induced podocyte dedifferentiation. Kidney Int 64:1695-1701, 2003.
22. He JC, Husain M, Sunamoto M, D'Agati VD, Klotman ME, Iyengar R, et al. Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1,2 pathways. J Clin Invest 114:643-651, 2004.
23. Dickie P, Roberts A, Uwiera R, Witmer J, Sharma K, Kopp JB. Focal glomerulosclerosis in proviral c-fms transgenic mice links Vpr expression to HIV-associated nephropathy. Virology 322:69-81, 2004.
24. Jowett JB, Planelles V, Poon B, Shah NP, Chen ML, Chen IS. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J Virol 69:6304-6313, 1995.
25. Zhong J, Zuo Y, Ma J, Fogo AB, Jolicoeur P, Ichikawa I, et al. Expression of HIV-1 genes in podocytes alone can lead to the full spectrum of HIV-1 associated nephropathy. Kidney Int 68:1048-1060, 2005.
26. Cohen AH, Sun NCJ, Shapshak P, Imagawa DT. Demonstration of human immunodeficiency virus in renal epithelium in HIV-associated nephropathy. Mod Pathol 1:87-97, 1989.
27. Bruggeman LA, Ross MD, Tanji N, Cara A, Dikman S, Gordon RE, et al. Renal epithelium is a previously unrecognized site of HIV-infection. J Am Soc Nephrol 11:2089-2087, 2000.
28. Marras D, Bruggeman LA, Gao F, Tanji N, Mansukhani MM, Cara A, et al. Replication and compartimentalization of HIV-1 in the kidney epithelium of patients with HIV-associated nephropathy. Nat Med 8:522-526, 2002.
29. Wintson JA, Bruggeman LA, Ross MD, Jacobson L, Ross L, D'Agati VD, et al. Nephropathy and establishment of a renal reservoir of HIV type 1 during primary infection. N Engl J Med 344:1979-1984, 2001.
30. Di Belgiosjoso GB, Genderini A, Vago L, Parravicini C, Bertoli S, Landriani N. Absence of HIV antigens in renal tissue from patients with HIV-associated nephropathy. Nephrol Dial Transplant 5:489-492, 1990.
31. Alpers CR, McClure J, Bursten SL. Human mesangial cells are resistant to productive infection by multiple strains of human immunodeficiency virus types 1 and 2. Am J Kidney Dis 19:126-130, 1992.
32. Eitener F, Cui Y, Hudkins KL, Anderson DM, Schmidt A, Morton WR, Alpers CE. Chemokine receptor (CCR5) expression in human kidneys and in the HIV infected macaque. Kidney Int 54:1945-1954, 1998.
33. Eitner F, Cui Y, Hudkins KL, Stokes MB, Segerer S, Mack M, et al. Chemokine receptor CCR5 and CXCR4 expression in HIV-associated kidney disease. J Am Soc Nephrol 11:856-867, 2000.
34. Ray PE, Bruggeman L, Weeks B, Kopp J, Bryant J, Owens J et al. Role of bFGF and its low affinity receptors in the pathogenesis of HIV-associated nephropathy in transgenic mice. Kidney Int 46:759-772, 1994.
35. Dickie P, Felser J, Eckhaus M, Bryant J, Silver J, Marinos N, et al. HIV-associated nephropathy in transgenic mice expressing HIV-1 genes. Virology 185:109-119, 1991.
36. Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, et al. Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc Natl Acad Sci USA 89:1577-1581, 1992.
37. Reid W, Sadowska M, Denaro R, Rao S, Foulke Jr J, Hayes N, et al. An HIV-1 transgenic rat that develops HIV-related pathology and immunology dysfunction. Proc Natl Acad Sci 98:9271-9276, 2001.
38. Ray PE, Liu XL, Robinson RL, Reid W, Xu L, Owens JW, et al. A novel HIV-1 transgenic rat model of childhood HIV-1 associated nephropathy. Kidney Int 63:2242-2253, 2003.
39. Bruggeman LA, Dikman S, Meng C, Quaggin SE, Coffman TM, Klotman PE. Nephropathy in human immunodeficiency virus-1 transgenic mice is due to renal transgene expression. J Clin Invest 100:84-92, 1997.
40. Tinkle BT, Ngo L, Luciw PA, Maciag T, Jay G. Human Immunodeficiency Virus-associated vasculopathy in transgenic mice. J Virol 71:4809-4814, 1997.
41. Tinkle BT, Ueda H, Ngo L, Luciw PA, Shaw K, Rosen CA, et al. Transgenic dissection of HIV-genes involved in lymphoid depletion. J Clin Invest 100:32-39, 1997.
42. Zietz C, Hotz B, Sturz M, Rauch E, Penning R, Lohrs U. Aortic endothelium in HIV-1 infection. Chronic injury, activation and increased leukocyte adherence. Am J Pathol 149:1887-1898, 1996.
43. Bussolino F, Mitola S, Serini G, Barillari G, Ensoli B. Interactions between endothelial cells and HIV-1. Int J Biochem Cell Biol 33:371-390, 2001.
44. Gilles PN, Lathey JL, Spector SA. Replication of macrophage-tropic and T-cell-tropic strains of Human Immunodeficiency Virus type 1 is augmented by macrophage-endothelial cell contact. J Virol 69:2133-2139, 1995.
45. Ascheri G, Sgadari C, Bugarini R, Bogner J, Schatz O, Ensoli B, et al. Serum concentration of fibroblast growth factor 2 are increased in HIV-1 type-infected patients and inversely correlated to survival probability. AIDS Res Hum Retroviruses 17:1035-1039, 2001.
46. Ray PE, Liu XH, Xu L, Rakusan T. Accumulation of bFGF in children with HIV-1 associated hemolytic uremic syndrome. Pediatr Nephrol 13:586-593, 1999.
47. Gharavi AG, Ahmad T, Wong RD, Hooshyar R, Vaughn J, Oller S, et al. Mapping a locus for susceptibility to HIV-1 associated nephropathy to mouse chromosome 3. Proc Natl Acad Sci USA 101:2488-2493, 2004.
48. Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science 300:1298-1300, 2003.
49. Gospodarowicz D, Ferrara N, Schweigerer L, Neufeld G. Structural characterization and biological functions of fibroblast growth factor. Endocrine Reviews 8:95-114, 1987.
50. Wellstein A, Czubayko F. Inhibition of fibroblast growth factors. Breast Cancer and Res Treatment 38:109-119, 1996.
51. Ensoli B, Gendelman R, Markham P, Fiorelli V, Colombinin S, Raffeld M, et al. Synergy between basic FGF and HIV-1 Tat protein in induction of Kaposi's sarcoma. Nature 371:674-680, 1994.
52. Li Z, Jerebtsova M, Liu XH, Tang P, Ray PE. Novel cystogenic role of basic fibroblast growth factor in developing rodent kidneys. Am J Physiol Renal Physiol 291:F289-F296, 2006.
53. Peoples GE, Blotnick S, Takahashi K, Freeman MR, Klasgsbrun M, Eberlein TJ. T lymphocytes that infiltrate tumors and atherosclerotic-plaques produce heparin-binding epidermal growth factor-like growth factor and basic fibroblast growth factor: a potential pathologic role. Proc Natl Acad Sci USA 95:6547-6551, 1995.
54. Celli G, LaRochelle J, Mackem S, Sharp R, Merlino G. Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J 17:1642-1655, 1998.
55. Clayton A, Thomas J, Thomas GJ, Davies M, Steadman R. Cell surface heparan sulfate proteoglycans control the response of renal interstitial fibroblasts to fibroblast growth factor-2. Kidney Int 59:2084-2094, 2001.
56. Sasaki T, Jyo Y, Tanda N, Kawakami Y, Nohno T, Tamai H, et al. Changes in glomerular epithelial cells induced by FGF-2 and FGF-2 neutralizing antibody in puromycin aminonucleoside nephropathy. Kidney Int 51:301-309, 1999.
57. Kriz W, Hahnel B, Rosener S, Elger M. Long term treatment of rats with FGF-2 results in focal segmental glomerulosclerosis. Kidney Int 48:1435-1450, 1995.
58. Morita H, Shinzato T, David G, Mizutani A, Habuchi H, Fujita Y, et al. Basic fibroblast growth factor-binding of heparan sulfate in the human glomerulosclerosis and renal tubulointerstitial fibrosis. Lab Invest 71:528-535, 1984.
59. Floege J, Kriz W, Schulze M, Susani M, Kerjaschki D, Mooney A, et al. Basic fibroblast growth factor augments podocytes injury and induces glomerulosclerosis in rats with experimental membranous nephropathy. J Clin Invest 96:2809-2819, 1995.
60. Naparsteck Y, Cohen IR, Fuks Z, Vlodavsky I. Activated T lymphocytes produce a matrix-degrading heparan sulphate endoglycosidase. Nature 252:241-244, 1984.
61. Bashkin P, Doctrow S, Klagsbrun M, Svahan CM , Folkman J, Vlodavsky I. Basic Fibroblast growth factor binds to subendothelial extracellular matrix and is released by heparitinase and heparin-like molecules. Biochemistry 28:1737-1743, 1989.
62. Tang P, Jerebtsova M, Przygodzki R, Ray PE. Fibroblast growth factor-2 increases the renal recruitment and attachment of HVI-infected mononuclear cells to renal tubular epithelial cells. Pediatr Nephrol 20:1708:1716, 2005.
63. Czubayko F, Liaudet-Coopman ED, Aigner A, Tuveson AT, Berchem GJ, Wellstein A. A secreted FGF-binding protein can serve as the angiogenic switch in human cancer. Nat Med 3:1137-1140, 1997.
64. Liu XH, Achim A, Xu L, Wellstein A, Ray PE. Up-regulation of a fibroblast growth factor binding protein in children with renal diseases. Kidney Int 59:1850-1858, 2001.
65. Ray PE, Liu XH, Henry D, Dye L, Xu L, Orenstein JM, et al. Infection of human primary renal epithelial cells with HIV-1 from children with HIV-associated nephropathy. Kidney Int 53:1217-1229, 1998.
66. Conaldi PG, Biancone L, Botelli A, Wasde-Evans A, Racusen LC, Boccellino M, et al. HIV-1 kills renal tubular epithelial cells in vitro by triggering an apoptotic pathway involving caspase activation and fas upregulation. J Clin Invest 102:2041-2049, 1998.
67. Ray PE, Garcia Soler A, Xu L, Soderland C, Blumenthal R, Puri A. Fusion of HIV-1 envelope expressing cells to human glomerular endothelial cells through a CXCR4 mediated mechanism. Pediatr Nephrol 20:1401-1409, 2005.
68. Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 co-receptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 17:657-700, 1999.
69. Liu XH, Hadley TJ, Xu X, Peiper SC, Ray PE. Up-regulation of Duffy antigen receptor expression in children with renal disease. Kidney Int 55:1491-1500, 1999.
70. Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson PJ, et al. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nature Med 6:769-775, 2000.
71. O'Donnell MP, Chao CC, Gekker G, Modi KS, Kasiske BL, Keane WF. Renal cell cytokine production stimulates HIV-1 expression in chronically HIV-infected monocytes. Kidney Int 53:593-597, 1998.
72. Patel M, Yanagishita M, Roderiquez G, Bou-Habib DC, Oravecz T, Hascall VC, et al. Cell-surface heparan sulfate proteoglycans mediates HIV-1 infection of T-cell lines. AIDS Res Hum Retroviruses 9:167-174, 1993.
73. Roderiquez G, Oravecz T, Yanagishita M, Bou-Habbi DC, Mostowski H, Norcross MA. Mediation of Human Immunodeficiency Virus-type 1 binding by interaction of cell surface Heparan Sulfate Proteoglycans with the V3 region of envelope gp120-gp41. J Virol 69:2233-2239, 1995.
74. Mondor I, Ugolini S, Sattentau QJ. Human immunodeficiency virus type 1 attachment to HeLa CD4 Cells is CD independent and gp120 dependent and requires cell surface heparan sulfate proteoglycans. J Virol 72:3623-3634, 1998.
75. Moulard M, Lortat-Jacob H, Mondor I, Roca G, Wyatt R, Sodroski J, et al. Selective interactions of polyanions with basic surfaces on HIV-1 gp120. J Virol 74:1948-1960, 2000.
76. Saphire ACS, Bobardt MD, Zhang Z, David G, Gallay PA. Syndecans serve as attachment receptors for Human Immunodeficiency virus type 1 on macrophages. J Virol 75:9187-9200, 2001.
77. Argyris EG, Acheampong E, Nunnari G, Mukhtar M, Williams KJ, Pomerantz RJ. Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J Virol 77:12140-12151, 2003.
78. Oravecz T, Pall M, Wang J, Roderiquez G, Ditto M, Norcross MA. Regulation of anti-HIV-1 activity of RANTES by heparan sulfate proteoglycans. J. Immunol 159:4587-4592, 1997.
79. Ibrahim J, Griffin P, Coombe DR, Rider CC, James W. Cell-surface heparan sulfate facilitates human immodeficiency virus Type 1 entry into some cells lines but not primary lymphocytes. Virus Res 60:159-169, 1999.
80. Bobardt MD, Saphire HC, Yu X, Schueren Van Der, Zhang Z, David G, et al. Syndecan captures, protects, and transmits HIV to T lymphocytes. Immunity 18:27-39, 2003.
81. Gordon CJ, Muesing MA, Proudfoot AE, Power CA, Moore JP, Trkola A. Enhancement of HIV-type 1 infection by the CC-chemokine RANTES is independent of the mechanism of virus-cell fusion. J Virol 73:684-694, 1999.
82. Schols D, Struyf S, Van Damme J, Este JA, Henson G, De Clercq E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J Exp Med 186:1383-1343, 1997.
83. Albini A, Ferrini S, Benelli R, Sforzini S, Giunciuglio D, Aluigi MG, Proudfoot AEI, et al. HIV-1 Tat protein mimicry of chemokines. Proc Natl Acad Sci USA 95:13153-13158, 1998.
84. Tyago M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 Tat requires Cell surface heparan sulfate proteoglycans. J Biol Chem 276:3254-3261, 2001.
85. Rusnati M, Tulipano G, Urbinati C, Tanghetti E, Giuliani R, Giacca M, et al. The Basic Domain in HIV-1 Tat protein as a target for polysulfonated heparin-mimicking extracellular Tat antagonist. J Biol Chem 273(26):16027-16037, 1998.
86. Bieniasz PD, Grdina TA, Bogerd HP, Culen BR. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J 23:7056-7065, 1998.
87. Westendorp MO, Frank R, Ochsenbauer C, Sticker K, Dhein J, Walczak H, et al. Sensitization of T cells to CD95-mediated apoptosis by HIV-Tat and gp120. Nature 375:497-500, 1995.
88. Jia H, Lohr M, Jezequel S, Davis D, Shaikh S, Selwood D, et al. Cysteine-rich and basic domain HIV-1 Tat peptides inhibit angiogenesis and induce endothelial cell apoptosis. Biochem Biophy Res Comm 283:469-479, 2001.
89. Boykins RA, Mahieux R, Shankavaram UT, Gho YS, Lee SF, Hewlett IK, et al. A short polypeptide domain of HIV-1 Tat proteins mediates pathogenesis. J Immunol 163:15-20, 1999.
90. Barillari G, Sgadari C, Fiorelli V, Samaniego F, Colombini S, Manzari V, et al. The Tat protein of human immunodeficiency virus type 1 promotes vascular cell growth and locomotion by engaging the a5 b1 and a v??b3 integrins and by mobilizing sequestered basic-FGF. Blood 94:663-672, 1999.
91. Toschi E, Barillari G, Sgadari C, Bacigalupo I, Cereseto A, Carlei D, et al. Activation of matrix-metalloproteinase-2 and membrane-type-1-matrix-metalloproteinase in endothelial cells and induction of vascular permeability in vivo by human immunodeficiency virus-1 Tat protein and basic fibroblast growth factor. Mol Biol Cell 12:2934-2946, 2001.
92. Mohan R, Sivak J, Ashton P, Russo LA, Pham BQ, Kasahara N, et al. Circuminoids inhibit the angiogenic response stimulated by fibroblast growth factor-2, including expression of matrix metalloproteinase gelatinase B. J Biol Chem 275:10405-10412, 2000.
93. Takeuchi A, Yoshizawa N, Yamamoto M, Sawasaki Y, Oda T, Senoo A, et al. Basic Fibroblast Growth Factor promotes proliferation of rat glomerular visceral epithelial cells in vitro. Am J Pathol 141:107-116, 1992.
94. Sasaki T, Hatta H, Osawa G. Cytokines and podocyte injury: the mechanism of fibroblast growth factor-2 induced podocyte injury. Nephrol Dial Transpl 14:33-34, 1999.
95. Szczech LA, Edwards LJ, Sanders LL, Van der Horst C, Bartlett JA, Heald AE, et al. Protease inhibitors are associated with a slowed progression of HIV-related renal diseases. Clin Nephrol 57:336-341, 2002.
96. Schwartz EJ, Szczech LA, Ross MJ, Klotman ME, Winston JA, Klotman PE. Highly active antiretroviral therapy and the epidemic of HIV+ end stage renal disease. J Am Soc Nephrol 16:2412-2420, 2005.
97. Wei A, Burns GC, Williams BA, Mohammed NB, Sivak SL. Long term renal survival in HIV-associated nephropathy with angiotensin-converting enzyme inhibition. Kidney Int 64:1462-1471, 2003.
98. Kumar MS, Sierka DR, Damask AM, Fyfe B, McAlack RF, Heifets M, Moritz MJ, et al. Safety and success of kidney transplantation and concomitant immunosuppression in HIV-positive patients. Kidney Int 67:1622-1629, 2005.
99. Bhagani S, Sweny PI, Brook K, British HIV association. Guidelines for kidney transplantation in patients with HIV disease. HIV Medicine 7:133-139, 2006.
|