Introducción
La tuberculosis (TBC) es una epidemia global que ha emergido nuevamente y se complica debido a la epidemia actual del síndrome de inmunodeficiencia adquirida (sida)/virus de la inmunodeficiencia humana (HIV) y por el uso de drogas inmunosupresoras. Esta enfermedad puede comprometer cualquier sistema del organismo y en el abdomen puede remedar una variedad de afecciones tales como enfermedad intestinal inflamatoria, neoplasias y otras enfermedades infecciosas.
Las características radiológicas, aunque no son patognomónicas, son muy sugestivas de la enfermedad cuando se las considera junto con la presentación clínica, el estado inmunológico y el perfil demográfico. Debido a la amplia variación en la presentación de la tuberculosis abdominal y a las diferentes modalidades del diagnóstico por imágenes, es necesario extraer mediante un formato basado en las pruebas existentes los aspectos importantes de cada modalidad que podrían ser útiles en la práctica radiológica. Esto es lo que propone esta revisión y el texto se divide según las áreas comprometidas dentro del abdomen:
peritoneo,
sistema linfático,
tracto gastrointestinal,
órganos sólidos.
La tuberculosis genitourinaria es una entidad separada, por lo cual se excluye de esta revisión.
Tuberculosis peritoneal
La peritonitis es una manifestación infrecuente de la tuberculosis, que se produce en menos del 4% de los pacientes y se presenta con distensión abdominal, dolor y fiebre. Aproximadamente el 15% de los casos se asocia con TBC pulmonar activa3,4 y se considera que la mayoría es consecuencia de la reactivación de focos latentes de TBC en el peritoneo, establecidos previamente a causa de la diseminación hematógena de un foco primario pulmonar. Además, la peritonitis tuberculosa puede acontecer luego de la diseminación hematógena de los sitios con infección primaria activa cercanos o remotos o por la liberación del material caseoso de un ganglio linfático afectado, de un segmento intestinal o de las trompas de Falopio. 2,5-8
Características generales
Tradicionalmente se describen tres tipos de peritonitis tuberculosa: “ascítico- húmeda”, “fibrótica-fija” y “plástica y seca”. El tipo “ascítico húmedo” es el más común y se caracteriza por grandes cantidades de líquido ascítico libre o tabicado.
La peritonitis “fibrótica-fija” es menos frecuente y se distingue por el compromiso del epiplón, un plastrón de asas intestinales y mesenterio y a veces por ascitis tabicada. La “plástica y seca” es inusual y se caracteriza por nódulos caseosos, reacción peritoneal fibrótica y adherencias densas.2,12-16 Sin embargo, esta clasificación no parece ser lo suficientemente precisa para reflejar todas las combinaciones de características radiológicas demostradas mediante las modalidades del diagnóstico por imágenes, debido a que el peritoneo, el epiplón mayor y el mesenterio del intestino delgado están sujetos a distintos grados de compromiso durante el transcurso de la enfermedad.17,18
El tipo “húmedo”, que se observa en el 90% de los casos, se caracteriza por grandes cantidades (el 97% de los pacientes tienen ascitis en el momento de su presentación3,4,19) de líquido ascítico viscoso que se distribuye en forma difusa o está tabicado en bolsillos complejos.13,15 El tipo “fibrótico y fijo” se manifiesta en el 60% de los casos y en la tomografía computada (TC) y en la ecografía (ECO) se caracteriza por la presencia de masas en el epiplón mayor, un plastrón de asas intestinales y mesenterio y ocasionalmente por ascitis tabicada.15 La peritonitis “plástica” o “seca” se presenta en el 10% de los casos y se identifica por los nódulos caseosos, la reacción peritoneal fibrótica y las adherencias densas.15,16 Estos signos sugieren tuberculosis pero no son específicos debido a que la diseminación de la neoplasia peritoneal, el mesotelioma y la peritonitis pueden ser similares en apariencia. Detectamos peritonitis en el 77% de los pacientes de nuestro estudio en los el tipo “húmedo” y “seco” eran los más comunes.20 Las complicaciones de la peritonitis tuberculosa comprenden la formación de abscesos interasa, que en algunas ocasiones son detectados únicamente mediante la TC, o bandas de adhesión que producen obstrucción. En la enfermedad avanzada la pérdida de planos adiposos puede ser la causa de la falta de definición de diversas estructuras anatómicas dentro del abdomen.
Características específicas
Ascitis
La ascitis libre o tabicada se manifiesta en 30% a 100% de los casos y se puede demostrar mediante la ECO o la TC.18,21,22 Sin embargo, la ECO se recomienda en la investigación inicial para visualizar pequeńas cantidades de líquido ascítico libre o tabicado que no se sospechan clínicamente (figura 1). Las características de la ECO que sugieren peritonitis tuberculosa son la presencia de bandas delgadas y fibrosas en el líquido ascítico, la ascitis localizada y los nódulos linfáticos caseosos o calcificados. Los restos ecogénicos visibles en la ECO representan la presencia de bandas finas de material particulado en el líquido ascítico que se observan en el 10% al 40% de los casos.13,15-17,23 La ECO permite delinear los septos múltiples, móviles y delgados y los restos de la ascitis en 10% a 100% de los pacientes (figuras 2, 3a).13,15-18,21,24- 26 En estudios retrospectivos de pacientes con ascitis, los tabiques se registran en el 10% al 72%,13,15-17 mientras que en ensayos prospectivos estos resultados oscilan entre 30% y 100%.21,24,27
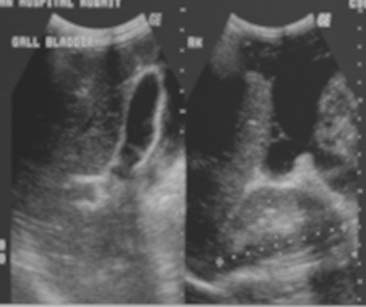
Figura 1
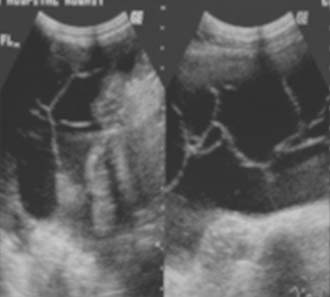
Figura 2
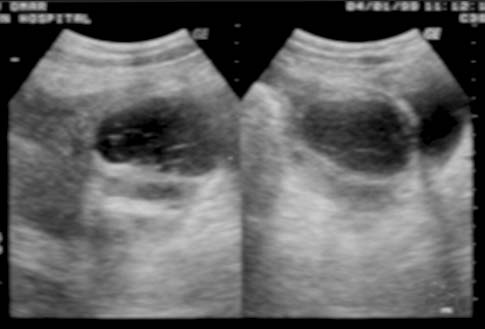
Figura 3a
En la TC, el líquido ascítico presenta valores altos de atenuación (20-45 UH) en la mayoría de los enfermos9,10,15,18,25,26 debido al contenido elevado de células y proteínas. La TC simple y con contraste demuestra claramente el nivel líquido/grasa en el líquido ascítico.28 El líquido ascítico de la TBC presenta valores bajos de atenuación cercanos a la densidad del agua en algunos sujetos,9,15,29 lo que probablemente refleja una fase trasudativa previa por la reacción inmune.18,29 Con la TC pueden encontrarse fracasos en cuanto a la detección de tabiques interconectados, múltiples y delgados en la mayoría de los pacientes, en especial en las regiones subdiafragmática y pelviana (figura 3b). En un estudio retrospectivo acerca de las características de la TC en la TBC abdominal encontramos grandes volúmenes de líquido ascítico en el 52% de los sujetos.20
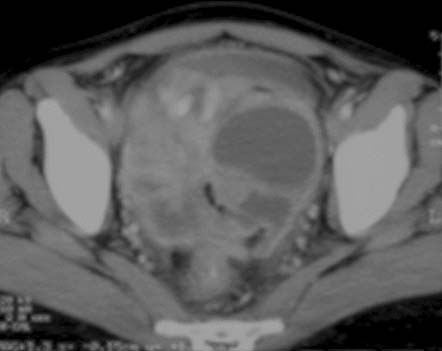
Figura 3b
Peritoneo
El engrosamiento del peritoneo y los nódulos pequeńos se visualiza mejor ante la presencia de ascitis, tanto en la ECO (figuras 4a y 4b) como en la TC (figuras 5, 6a y 6b); sin embargo, los cambios peritoneales no fueron evaluados o descritos separadamente en la mayoría de los estudios que emplearon ECO.13,15,16,22,27 El engrosamiento peritoneal difuso, regular o irregular hipoecoico con nódulos pequeńos o sin éstos puede ser detectado mediante la ECO17 (figura 4).21,24 El peritoneo se encuentra engrosado en alrededor del 14% al 100% de los pacientes con ascitis.8,17,21,24 La aparición de engrosamiento peritoneal se describió inicialmente en siete pacientes con ascitis, de los cuales tres presentaron el realce del peritoneo luego de la administración de contraste.18 Desde entonces, el engrosamiento leve y uniforme del peritoneo o el realce pronunciado con el contraste se informó en la mayoría de los casos en presencia de ascitis.15,24,29,30 La TC es la mejor modalidad para el diagnóstico por imágenes. Revela masas moteadas de baja densidad o el engrosamiento nodular de tejido blando a lo largo de las superficies peritoneales más vascularizadas, del mesenterio y del epiplón, como también las asas intestinales apelmazadas26 (figura 6).15,23
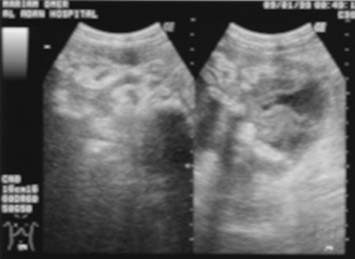
Figura 4a
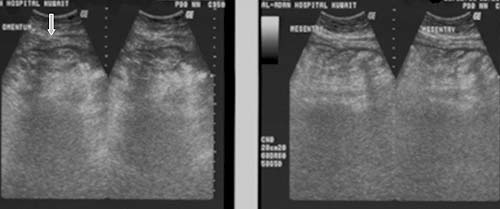
Figura 4b
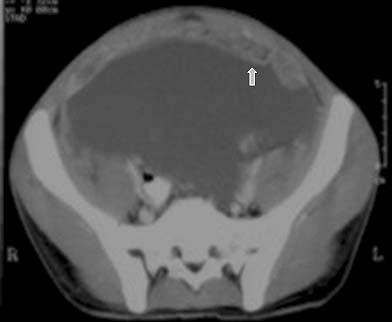
Figura 5

Figura 6a
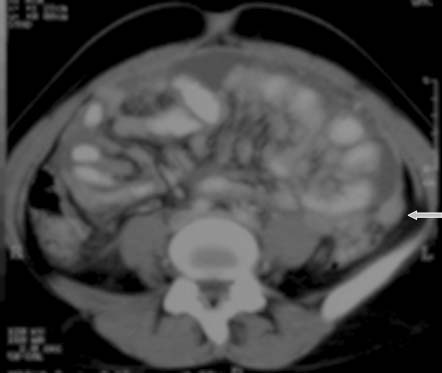
Figura 6b
Epiplón
La localización superficial del epiplón mayor permite que sea fácilmente diferenciado mediante las modalidades del diagnóstico por imágenes. El compromiso del epiplón puede tener apariencia “nodular”, “manchada” o “apelmazada” y se demostró en alrededor del 14% al 55% de los casos, según distintas series.13,16,24 El engrosamiento nodular se observa en la ECO en ocasiones (figura 4) como el engrosamiento del epiplón con ecogenicidad semejante a la de una hoja, asas intestinales engrosadas y adheridas con exudado o ascitis escasa que le otorgan el aspecto en “rebanada de pan” y engrosamiento mesentérico con áreas ecogénicas bizarras e hipoecoicas13,16,31,32 que se asemejan a un “emparedado de helado”.25 La TC aumenta la detección de cambios en el epiplón en alrededor del 36% al 82%.9,15,22,24,30,33 El compromiso de tipo moteado en la TC es el más común en sujetos con compromiso del epiplón30 (figuras 5 y 6).33 En la resonancia magnética nuclear (RMN) es habitual el engrosamiento del epiplón con ascitis y la presencia de una masa pelviana bilateral.7
Mesenterio del intestino delgado
Como parte integral del peritoneo, el mesenterio del intestino delgado también se encuentra afectado en la peritonitis tuberculosa. Los cambios mesentéricos más comunes son las lesiones nodulares, el engrosamiento del mesenterio y la pérdida de la configuración normal de este último.33 Las lesiones mesentéricas nodulares pueden presentarse como micronódulos (< 5 mm) o macronódulos (> 5 mm) sólidos o quísticos en los ganglios linfáticos o abcesos.18,33 En la ECO y la TC las asas intestinales fijas y el mesenterio se presentan como rayos que irradian desde la raíz del mesenterio, denominado signo estrellado (figura 7a y 7b). En la ECO se observa el engrosamiento del mesenterio (> de 15 mm) con ganglios linfáticos mesentéricos y se describe esta combinación como un signo temprano de TBC abdominal.27 El engrosamiento ecográfico del mesenterio con ganglios linfáticos o sin ellos también se describió en distintas series.13,17,24 En nuestra experiencia, el engrosamiento peritoneal, mesentérico o del epiplón y la formación de masas (peritonitis seca) se observó en el 44% de los casos.20 La enfermedad mesentérica se demostró en la TC como un aumento de la vascularización y por bandas engrosadas dentro del mesenterio, que se agrupan entre sí por la inflamación, con asas intestinales apelmazadas que forman una masa abdominal (figura 8). Esta masa también puede formarse por la ascitis, los abcesos tabicados y los ganglios linfáticos.15 El signo estrellado que representa el engrosamiento y rigidez del mesenterio, el compromiso de tipo nodular y el patrón nodular o la pérdida de la configuración normal del mesenterio se detectan mejor con la TC10 que con la ECO15 (figura 7).18,24,30,33
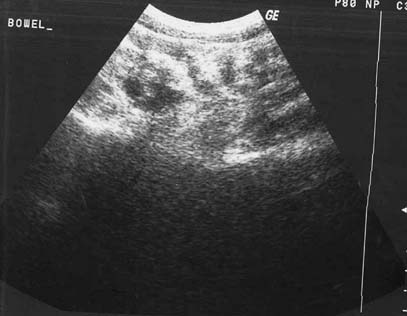
Figura 7a
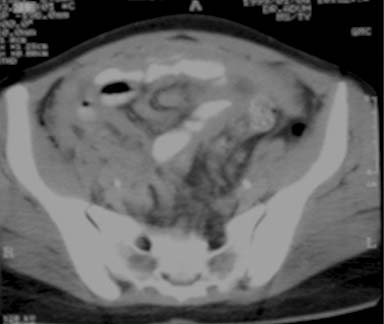
Figura 7b

Figura 8
Diagnóstico diferencial de la peritonitis tuberculosa
Como el peritoneo y sus extensiones constituyen distintas partes integrales, las características radiológicas de la peritonitis tuberculosa pueden imitar las observadas en diversas enfermedades. El diagnóstico diferencial más importante debe incluir carcinomatosis peritoneal, mesotelioma peritoneal primario, peritonitis y, con menor frecuencia, linfoma.17,21,26,29,30,34 Dos estudios publicados recientemente intentaron determinar la utilidad de la TC para diferenciar la TBC peritoneal de la carcinomatosis peritoneal.30,33
El peritoneo uniforme con engrosamiento mínimo y que se realza en forma pronunciada con el contraste sugiere TBC peritoneal, mientras que las siembras nodulares y el engrosamiento irregular del peritoneo sugieren carcinomatosis peritoneal.30 Además, el compromiso del mesenterio asociado con los macronódulos (mayores de 5 mm de diámetro), una línea fina en el epiplón (una pared fibrosa que cubre el epiplón infiltrado), masas peritoneales o extraperitoneales con centros hipodensos y calcificación y la esplenomegalia o calcificaciones esplénicas se observaron con mayor frecuencia en la peritonitis tuberculosa.33 A su vez, la inflamación que se extiende a lo largo del peritoneo hasta el compartimiento extraperitoneal sugiere TBC. La sensibilidad global de la TC para predecir la peritonitis tuberculosa es del 69% y del 91% para la carcinomatosis peritoneal.33 Las características de la peritonitis tuberculosa, como el engrosamiento peritoneal, el compromiso del epiplón y del mesenterio y la formación de tabiques en la ascitis, también se encontraron en los pacientes con mesotelioma peritoneal. Los signos sugestivos de mesotelioma peritoneal son el engrosamiento peritoneal multifocal semejante a una hoja o el tipo nodular con nódulos de hasta 3 cm, masas de tejido blando mesentéricas, del epiplón o del peritoneo, tabiques gruesos y rígidos entre las hojas del peritoneo, asas intestinales fijas y ascitis escasa que no es proporcional al grado de diseminación tumoral. Mientras que las características que sugieren peritonitis tuberculosa son la presencia de engrosamiento peritoneal mínimo y uniforme (< 5 mm) que se realza en forma pronunciada con el contraste y tabiques múltiples, finos, completos o incompletos y móviles.21,24,33,34 El uso de la RMN es limitado, pero el realce que se obtiene 15 a 20 minutos después de la administración de Gd-DTPA por vía endovenosa no es una característica infrecuente en la ascitis exudativa, aunque este signo se registró en 1 de 2 pacientes con peritonitis tuberculosa.35
Tuberculosis de los ganglios linfáticos
La linfadenopatía tuberculosa es la manifestación más común de la TBC abdominal que puede ser transmitida por medio de tres vías principales. La primera es la ingestión de material infectado con bacilos tuberculosos, como el esputo o la leche.
Debido a que el drenaje desde los linfáticos ubicados del lado izquierdo del colon hacia los ganglios linfáticos mesentéricos inferiores en L3 es raro, es infrecuente que los ganglios linfáticos paraaórticos inferiores estén comprometidos. El compromiso tuberculoso del duodeno no es inusual y los bacilos son transportados desde los ganglios linfáticos del duodeno hacia los del ligamento hepatoduodenal y la región peripancreática. El drenaje linfático desde el intestino explica el compromiso de los ganglios mesentéricos, pararrenales anteriores, paraaórticos superiores y, en menor medida, de los ganglios linfáticos del epiplón, con excepción de los ganglios paraaórticos inferiores. La linfadenopatía retroperitoneal es infrecuente, la mayoría de los pacientes con compromiso de los ganglios linfáticos retroperitoneales presentan compromiso ganglionar en otros sitios. La segunda vía de transmisión es la diseminación hematógena. Las bacterias se propagan a partir de sitios infectados distantes, habitualmente los pulmones, hacia el sistema linfático abdominal. Debido a que este proceso es sistémico, puede ocasionar infección del mesenterio, del epiplón menor, los ganglios linfáticos pararrenales anteriores y paraaórticos superiores e inferiores. La tercera vía de transmisión es la diseminación de la infección directamente a los ganglios linfáticos desde la serosa de las glándulas o de las estructuras adyacentes infectadas. La TBC de los órganos reproductivos puede diseminarse hacia los ganglios paraaórticos superiores e inferiores por el drenaje linfático.
Las características patológicas de los especímenes quirúrgicos de la linfadenopatía tuberculosa indican que las sustancias de caseificación y licuefacción en el centro de los ganglios linfáticos agrandados presentan baja atenuación y que presumiblemente son consecuencia de la irrigación insuficiente, mientras que la inflamación periférica del tejido linfático es resultado de la irrigación conservada.
Los ganglios linfáticos con TBC miden menos de 4 cm de diámetro, en promedio 2 cm, que coincide con los datos que indican que el crecimiento patológico es autolimitado. El agrandamiento ganglionar podría ser interpretado como un tumor peritoneal o retroperitoneal y conducir a la resección quirúrgica innecesaria. La frecuencia de adenopatías en los pacientes con TBC abdominal varía considerablemente en la literatura, registrándose en el 25% al 93% de los casos.17,36-41 En nuestros estudios osciló entre el 30% 25,42 y el 49%.20 En alrededor de la mitad de los pacientes constituye una manifestación aislada sin otros indicios de compromiso abdominal.18 La linfadenopatía tuberculosa se asocia habitualmente con TBC gastrointestinal y con menor frecuencia con compromiso del peritoneo o de los órganos macizos; sin embargo, puede ser el único signo de enfermedad, en especial en la región periportal.38,43,44 Debido a que la TBC puede afectar todas las regiones linfáticas del abdomen, la distribución de los ganglios linfáticos aumentados de tamańo refleja simplemente el drenaje linfático de los órganos comprometidos y depende del sitio en los casos individuales.38,41 En una serie de 49 pacientes examinados con TC observamos linfadenopatía en el 47% (de tipo difusa en el 48%, localizada en el mesenterio en el 28%, peripancréatica y paraaórtica en el 13% cada una).20 Los sitios principalmente afectados por la linfadenopatía tuberculosa son las áreas periportales y peripancreáticas, pero en los pacientes con sida el compromiso puede ser más extenso. A pesar del volumen considerable de las adenopatías en algunos sujetos, no se informaron casos de obstrucción del tracto urinario o gastrointestinal. Sin embargo, puede aparecer obstrucción biliar secundaria a la compresión ductal directa por los ganglios infectados en asociación con la inflamación y la estenosis periductal.45-47 Es infrecuente que la trombosis de la vena porta y la hipertensión portal se presenten como complicaciones de la TBC abdominal que compromete los ganglios linfáticos del hilio hepático.47
Características ecográficas
A pesar de que el gas intestinal puede evitar la visualización de los ganglios abdominales,25 la ECO permite visualizarlos con frecuencia, en especial los grupos paraaórticos, paracava y mesentéricos.37 En la ECO, los ganglios linfáticos pueden ser discretos o presentarse como masas opacas en conglomerados. Es habitual que los ganglios agrandados contengan áreas centrales hipoecoicas37 (figuras 9a y 9b).16,17,39 En algunos pacientes pueden observarse calcificaciones ganglionares.17
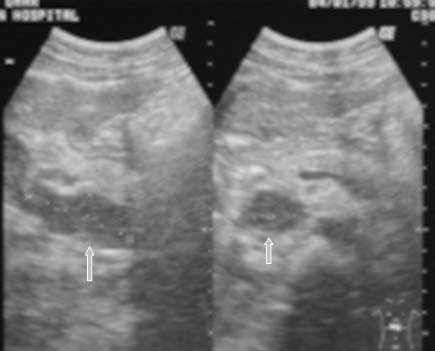
Figura 9a
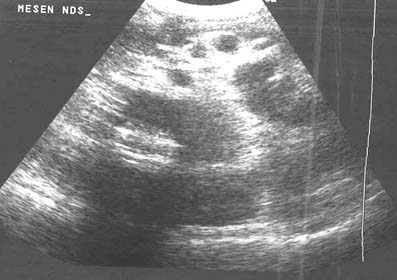
Figura 9b
Características en la TC
En la mayoría de los pacientes (40% al 70%), la TC muestra ganglios agrandados con centros hipodensos y márgenes periféricos hiperdensos que se realzan con el contraste37,48 (figuras 10a y 10b). Otros patrones de la morfología ganglionar en la TC comprenden: (i) masas ganglionares de densidad mixta en conglomerados, que podrían reflejar la presencia de ganglios múltiples confluentes secundarios a la diseminación perinodal de la inflamación (figura 11); (ii) ganglios agrandados de densidad homogénea, que con frecuencia se asocian con ganglios hipodensos en otros sitios, y (iii) aumento en el número (mayor de 3 en un corte de la TC) de ganglios mesentéricos de tamańo normal o ligeramente agrandados de densidad homogénea, habitualmente ubicados a lo largo de los vasos mesentéricos o adyacentes a las asas intestinales (figura 8). La atenuación en la TC de los tejidos centrales que no se realzan con el contraste oscila entre 28 y 84 UH y es compatible con sustancias de caseificación y licuefacción central. En la TC, estas características morfológicas diferentes pueden reflejar la evolución de los distintos estadios patológicos de la enfermedad, con granulomas sin caseificación al inicio seguidos de caseificación y necrosis.48 El aumento de tamańo de los ganglios linfáticos se evalúa mejor con la TC que con la ECO.37 En las imágenes sin contraste, los ganglios pueden presentar valores bajos de atenuación (< 30 UH) o valores de atenuación de tejidos blandos similares a los del músculo (> 35 UH).

Figura 10a

Figura 10b
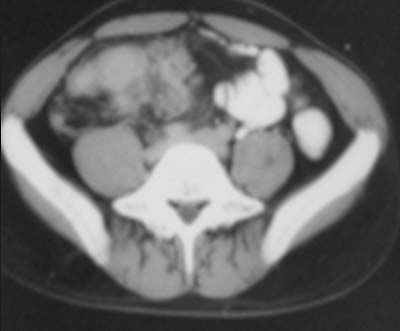
Figura 11
Se describen 4 patrones diferentes de agrandamiento ganglionar luego de la administración de contraste en los pacientes con tuberculosis.49 El patrón más común es el realce periférico, que se caracteriza por aumento del contraste en la periferia del ganglio con o sin puntos de baja atenuación en el centro. Los otros incluyen zonas que se realzan en forma heterogénea debido a la presencia de áreas focales dentro de los ganglios que no se destacan con el contraste, realce homogéneo y ganglios de baja atenuación que no captan el contraste que se observan únicamente en los pacientes con sida38 (figura 12).

Figura 12
Características de la RMN
Moon y col.50 informaron una correlación clinicopatológica con la apariencia de la linfaadenitis tuberculosa mediastinal en la RMN. En su estudio, la porción periférica que se realza con el contraste corresponde a la reacción vascular inflamatoria, mientras que la porción que no capta el contraste corresponde a la caseificación o necrosis con licuefacción, dentro de los ganglios. Otros informes describieron seńales de baja intensidad en las imágenes ponderadas en T1 y T2.51 La variabilidad de la intensidad de las seńales en T2 puede representar la presencia de radicales libres dentro de la lesión de acuerdo con el estadio de evolución. El patrón con contraste se caracteriza por el retraso en el relleno del margen periférico en estudios con RMN dinámica con apariencia multilocular o sin ella. La RMN es útil para diferenciar los ganglios linfáticos agrandados lindantes con el páncreas diagnosticados inicialmente como una neoplasia quística en la TC abdominal; en la RMN se observa un área central necrótica con márgenes bien marcados. La RMN sirve para identificar los ganglios linfáticos agrandados y su relación con la vía biliar o las estructuras vasculares adyacentes. A pesar de que el diagnóstico de TBC es posible si estos hallazgos se acompańan de una historia clínica con otros sitios afectados por la enfermedad, la diferenciación entre la patología biliar y pancreática puede ser difícil para detectar la linfadenopatía tuberculosa periportal localizada. La RMN es útil para identificar la presencia de ganglios linfáticos aumentados de tamańo y su relación con la vía biliar y las estructuras vasculares adyacentes.
Calcificación de los ganglios linfáticos
Con la eliminación de la TBC bovina como problema clínico en la mayoría de los países desarrollados, la incidencia de calcificación de los ganglios linfáticos disminuyó. Aun así, los ganglios linfáticos calcificados pueden encontrarse en una radiografía simple de abdomen en un paciente sin antecedentes de enfermedad intestinal. Es habitual que estos sujetos sean asintomáticos, pero pueden presentar dolor abdominal secundario a adherencias. La apariencia de un ganglio linfático tuberculoso calcificado puede variar, pero casi siempre exhibe las características morfológicas de una masa sólida. Los ganglios linfáticos calcificados difieren en tamańo. Unos pocos pueden alcanzar 7 cm en el eje longitudinal pero lo habitual es que oscilen entre 1 y 3 cm de diámetro. La calcificación distrófica de los ganglios linfáticos puede manifestarse en cualquier cadena ganglionar abdominal, pero por lejos los ganglios linfáticos mesentéricos son los más afectados y esto representa el efecto de la infección previa, en cada una de las etapas producidas por las micobacterias.52 Anteriormente, cuando la TBC intestinal era común, el compromiso de los ganglios linfáticos mesentéricos era casi universal.
Casi todos los pacientes que presentaban ganglios mesentéricos calcificados habían tenido infección tuberculosa pulmonar en la primera infancia. Además, no todos los que poseen ganglios linfáticos mesentéricos calcificados serán reactivos a la tuberculina. La calcificación no es patognomónica de la TBC y raramente se observa en las metástasis del teratoma testicular y en el linfoma no Hodgkin luego del tratamiento. No obstante, la calcificación ganglionar en individuos de áreas endémicas ante la ausencia de un tumor primario conocido sugiere la etiología tuberculosa, en especial si se acompańa de la distribución y apariencia características de los ganglios. En los adultos, sólo unas pocas patologías diferentes de la tuberculosis dan lugar a calcificaciones ampliamente distribuidas en el abdomen. El quiste hidatídico, que se forma presumiblemente a partir de la ruptura de un quiste hepático primario dentro de la cavidad peritoneal, también puede calcificarse. Los flebolitos múltiples y pequeńos algunas veces pueden distribuirse ampliamente en la hemangiomatosis y comprometer el mesenterio, el intestino y el epiplón. Los quistes calcificados del mesenterio o del peritoneo, los quilosos en especial, pueden presentar calcificaciones uniloculares o multiloculares.
Tuberculosis en comparación con linfoma
La TC con contraste es útil para diferenciar la linfadenopatía abdominal tuberculosa de la linfadenopatía abdominal linfomatosa sobre la base de la anatomía y los patrones de captación de contraste en los ganglios linfáticos agrandados. La TBC diseminada y no diseminada afecta principalmente los ganglios del epiplón menor, los mesentéricos, pararrenales anteriores y paraaórticos superiores. Los ganglios linfáticos paraaórticos inferiores se encuentran comprometidos en el 5% de los pacientes con TBC no diseminada. En cambio, en los sujetos con linfomas, los más afectados son los ganglios linfáticos inferiores.
También se observa una diferencia distintiva entre los patrones ganglionares de captación de contraste; en la linfadenopatía tuberculosa predomina el patrón periférico y frecuentemente multilocular, en tanto que en el linfoma predomina el homogéneo.53 Sin embargo, en el 12.5% de los pacientes con enfermedad de Hodgkin y en el 26% de los sujetos con linfoma no Hodgkin existe una combinación del patrón homogéneo y del periférico con el contraste. Puesto que en los pacientes con linfoma los ganglios linfáticos agrandados poseen menor densidad y fijación al mesenterio luego del tratamiento mientras que en la recaída de la enfermedad, su apariencia es homogénea, es importante saber si los individuos recibieron tratamiento que puede ser la causa de la baja atenuación central dentro de los ganglios que remedan la linfadenopatía tuberculosa. En los pacientes con linfoma no Hodgkin, la distribución anatómica es semejante a la de los sujetos con TBC diseminada, no obstante, en los pacientes con enfermedad de Hodgkin, la distribución es diferente, ya que el compromiso de los ganglios mesentéricos es menos frecuente.
Tuberculosis del tracto gastrointestinal
La TBC que compromete el tracto gastrointestinal es rara y la incidencia informada en las internaciones hospitalarias es de 0.8%. La radiografía simple de tórax puede ser normal en 50% a 65% de estos pacientes.54-56 El diagnóstico radiológico tradicional se realiza sobre la base de las características en los estudios con bario.2,4,5 Actualmente, los hallazgos de la ECO, TC y las seriadas gastrointestinales (GI) se combinan para delinear la extensión total de la enfermedad. El diagnóstico puede obtenerse mediante la biopsia guiada por ECO o TC.57 Cualquier segmento del tracto GI puede estar comprometido en la TBC, pero la región ileocecal es afectada con mayor frecuencia y se registra en hasta el 90% de los casos con TBC intestinal.17,22,54,58 El compromiso de los ganglios linfáticos regionales es consecuencia de la diseminación linfática. Los ganglios regionales se adhieren a la pared intestinal enferma y forman una masa inflamatoria.2,54,58,59 En nuestra serie, la TBC del tracto GI se observó en el 38% de los casos y, de éstos, el 50% presentó enfermedad ileocecal.20
Tuberculosis esofágica
La TBC del esófago es una patología rara que se manifiesta por la propagación secundaria desde sitios adyacentes como los ganglios linfáticos, la TBC pulmonar y la vertebral.60,61 La ingestión de esputo infectado probablemente sea la causa de la TBC esofágica si están presente una úlcera mucosa o estenosis preexistentes. Puede ser secundaria a la diseminación linfática retrógrada60 y, con menor frecuencia, a la diseminación directa desde sitios adyacentes. La TBC esofágica primaria es muy infrecuente. Los exámenes con bario demuestran la compresión extrínseca de los ganglios linfáticos agrandados sobre el esófago, la fístulas esófago-bronquiales, formación de tractos sinuosos, irregularidad de la mucosa y ulceración. La TC es un método confiable para determinar la extensión total de la enfermedad en el mediastino.61
Gastritis y duodenitis tuberculosas
El compromiso gástrico y duodenal es muy raro y tiene lugar a causa de la diseminación secundaria desde los ganglios linfáticos adyacentes o por la vía hematógena, debido a la escasez de tejido linfático en dichas áreas. Cuando existe compromiso, el antro y el cuerpo gástrico distal son los sitios afectados con mayor frecuencia (figura 13). Las manifestaciones habituales comprenden úlceras, infiltración nodular de la pared gástrica, obstrucción de la salida del estómago o una masa gástrica. Las características radiológicas son inespecíficas e imitan los signos de la úlcera benigna, en la forma ulcerativa de la TBC, y las neoplasias, en la forma hipertrófica. Puede haber obstrucción de la salida debido a los cambios inflamatorios, la fibrosis o los ganglios linfáticos agrandados.2,15,62 El patrón de linitis plástica puede ser semejante al del carcinoma escirroso, sarcoidosis, sífilis, linfoma, ingestión previa de un agente cáustico o lesión por radiación. Por lo tanto, deben ser diferenciados radiológicamente. El patrón de erosión mucosa puede asemejarse a la gastritis corrosiva, la gastritis eosinofílica, la enfermedad de Crohn, la gastritis viral y la ingestión de un agente cáustico. El patrón similar a una masa puede remedar el carcinoma, leiomioma, leiomiosarcoma o sarcoma de Kaposi. La TC es muy importante para delinear la linfadenopatía regional. La presencia de un seno o de una fístula es rara pero sugiere TBC.15 Encontramos 2 casos de TBC gástrica y uno de ellos se presentó como una masa grande ulcerada y perforante.20
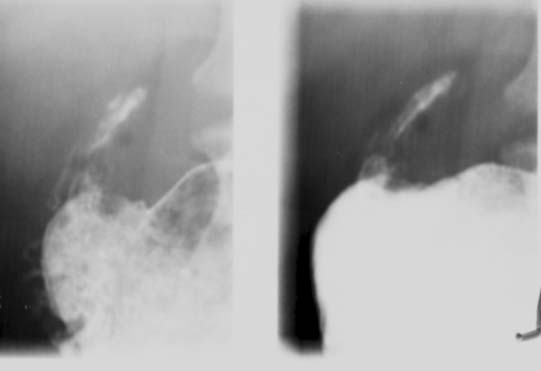
Figura 13
El compromiso duodenal es extremadamente raro y se presenta en hasta el 2% de los pacientes con TBC GI.22 La obstrucción duodenal se manifiesta como una estrechez y algunas veces como una fístula debido a los ganglios linfáticos adyacentes. La estenosis duodenal debida al aumento del tamańo ganglionar se observa en la tercera o la cuarta porción del duodeno. Mediante la ECO y la TC se puede demostrar una masa apelmazada que contiene los ganglios linfáticos agrandados y la raíz del mesenterio engrosada.15,18 El compromiso duodenal generalmente produce estenosis debido a la linfadenopatía adyacente.15 Se han descrito ejemplos aislados de infiltración directa del duodeno, que en la TC se manifiesta por el aumento del tamańo de las asas duodenales, con engrosamiento de sus capas y un tracto fistuloso4 o el engrosamiento mucoso inespecífico.26
Enteritis tuberculosa
El compromiso tuberculoso del intestino delgado aparte del íleon terminal es infrecuente y es habitual que se asocie con peritonitis. La TC y los estudios con bario demuestran cambios inespecíficos.12 En los estudios con bario se observan el engrosamiento de la capa mucosa (figura 14) y los sitios exactos de estenosis con dilatación proximal prominente del intestino delgado (figuras 15). Las úlceras no estenóticas que se caracterizan radiológicamente por una mancha de bario rodeada por el engrosamiento de la pared o por pliegues convergentes también pueden encontrarse en algunos pacientes con TBC del intestino delgado.
En dos casos se informó la presencia de estenosis yeyunal única que abarcaba la circunferencia junto con una úlcera circundante en los estudios de doble contraste.63 En las imágenes de la TC, el engrosamiento de la pared intestinal puede observarse junto con el engrosamiento del mesenterio, la fijación y la linfadenopatía que lleva a la separación de las asas intestinales12 (figuras 16a y 16b). Puede producirse neumatosis intestinal en los pacientes con TBC abdominal como consecuencia de la isquemia debido al compromiso mesentérico o a la enfermedad pulmonar asociada.
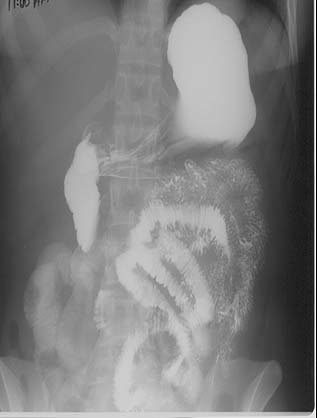
Figura 14

Figura 15
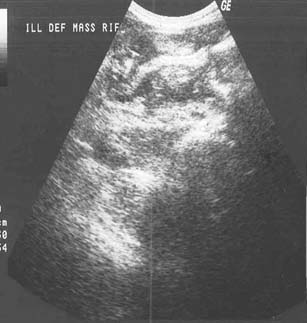
Figura 16a
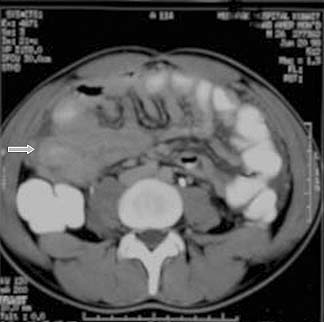
Figura 16b
Tuberculosis ileocecal y colónica
El compromiso del tracto gastrointestinal en la TBC se produce en la región ileocecal en el 90% de los casos.1,54 Esto se relaciona con la abundancia de tejido linfático y con el estasis relativo. No obstante, se describió el compromiso de todas las secciones del colon.64,65 La tuberculosis ileocecal se detectó en el 50% de nuestros pacientes con TBC del tracto GI.30
Estudios con bario
El compromiso inicial de la región ileocecal se manifiesta en el estudio de bario simple como el espasmo y la hipermotilidad con edema de la válvula. El engrosamiento de las valvas de la válvula ileocecal o el ensanchamiento del espacio valvular con estrechez del íleon terminal (signo de Fleischner) se describieron como características de la TBC.1 El detalle de la mucosa se logra por medio de los exámenes con bario de doble contraste que permiten la visualización de la ulceración en etapas tempranas de la enfermedad. También se comunicó la presencia de ulceración aftosa semejante a la observada en la enfermedad de Crohn, que se manifiesta como manchas de bario punteadas y superficiales rodeadas por un halo translúcido en el centro de la mucosa normal.64,66 El examen con bario revela el engrosamiento de las capas y la irregularidad del contorno.54 También se informaron lesiones salteadas en el íleon.54,67
En la enfermedad avanzada las deformidades características comprenden la estenosis anular simétrica “en servilletero” y la obstrucción, acortamiento, retracción y formación de bolsillos.15,67 El ciego se vuelve cónico, se contrae y se retrae fuera de la fosa ilíaca debido a la contracción del mesocolon.
El ángulo hepático puede ser tironeado hacia abajo por el mismo mecanismo.67 También pueden observarse nódulos submucosos y polipoides con intraluminales.65,67 La válvula ileocecal se vuelve fija, irregular, entreabierta e incompetente. El íleon terminal puede estar fijo y estrecho debido a la formación de estenosis (figura 15). El íleon terminal puede estrecharse debido a la irritabilidad con vaciamiento rápido del segmento enfermo a través de la válvula ileocecal entreabierta dentro del ciego acortado, rígido u obliterado (signo de Stierlin). Esto representa la inflamación aguda que se superpone en un segmento con compromiso crónico. A medida que progresa la enfermedad, pueden aparecer fisuras profundas y fístulas.
Características ecográficas
La ecografía es capaz de detectar los cambios producidos por la TBC intestinal en más del 90% de los casos.68 El intestino enfermo se reconoce por el engrosamiento inespecífico de su pared (un halo hipoecoico que mide más de 5 mm). El patrón de la enfermedad es difuso en el 80% de los casos y concéntrico en el 75%.68 En la mayoría de los pacientes la ecografía generalmente demuestra el engrosamiento uniforme y concéntrico de la pared intestinal (figura 16a). En ocasiones se observa ulceración.12,17 También pueden identificarse por la ECO las masas opacas constituidas por las asas intestinales y el peritoneo engrosados, la ascitis y los ganglios regionales.12,13,17 Las masas complejas con hallazgos asociados como ascitis, linfadenopatía y un epiplón apelmazado, aunque son inespecíficos de la TBC intestinal, también pueden observarse con la progresión de la enfermedad; la localización de la enfermedad en el cuadrante inferior derecho es sugerente.17,68
Características en la tomografía computada
La TC es una modalidad importante del diagnóstico por imágenes debido a que permite identificar cambios luminales y también características extraluminales accesorias como las adenopatías y los cambios mesentéricos (figura 16b); sin embargo, los estudios con bario son esenciales para caracterizar la naturaleza de la patología intestinal en cada paciente.15,18,54 Las características radiológicas son similares a las de la enfermedad de Crohn, linfoma, amebiasis, carcinoma e incluso sarcoidosis.2 La diferenciación radiológica de las etapas tempranas de la TBC ileocecal con la enfermedad de Crohn y el linfoma es habitualmente imposible; no obstante, las características en la TC de la TBC ileocecal avanzada no son compatibles con la enfermedad de Crohn y son inusuales en el linfoma ileocecal.54 Se acepta que la TC es capaz de identificar cambios en la pared intestinal y el mesenterio, lo que provee algunos factores de diferenciación entre la TBC intestinal y la enfermedad de Crohn. En la TBC los cambios de la pared intestinal que varían y reflejan las distintas etapas de la enfermedad son las masas exofíticas de tejido blando que rodean la luz comprimida y ulcerada, el engrosamiento mínimo y asimétrico de la pared con el contorno mucoso puntiagudo y fijo, el engrosamiento mínimo y simétrico de la pared y la ausencia de engrosamiento de la pared. La estratificación mural no se manifiesta en estos casos. En cambio, en la enfermedad de Crohn el engrosamiento de la pared es uniforme, concéntrico y simétrico y oscila entre 0.6 y 1.7 cm. Algunos de los pacientes presentan estratificación mural.58 Si bien la amebiasis produce el acortamiento típico del ciego que se observa en la TBC, la asociación de compromiso del intestino delgado es rara en la amebiasis. El carcinoma cecal siempre está limitado por la válvula ileocecal.
La apendicitis tuberculosa
La tuberculosis limitada al apéndice es infrecuente, no se asocia con ninguna característica clínica particular, se la confunde habitualmente con la apendicitis y la enfermedad inflamatoria pelviana y puede conducir a la perforación con la consiguiente diseminación del patógeno. En consecuencia, se sugiere la apendicectomía en todos los pacientes con tuberculosis abdominal que son sometidos a una laparotomía.
Organos sólidos
Páncreas
La TBC pancreática es extremadamente rara y sólo se comunicaron pocos casos en la bibliografía.4,18,69-71 A pesar de que el páncreas se compromete en la TBC miliar, puede ser el único sitio de reactivación de la TBC luego de muchos ańos.71 Las lesiones tuberculosas en el páncreas suelen estar localizadas en la cabeza y con menor frecuencia en el cuerpo y la cola. En la ECO las lesiones de la tuberculosis pancreática solitarias se visualizan como una masa bien definida hipoecoica y en la TC como una masa expansible e hipodensa de márgenes irregulares con o sin agrandamiento de todo el páncreas; la periferia de la lesión puede realzarse luego de la inyección de contraste endovenoso.15,22,70 Es infrecuente que la ECO y la TC muestren un agrandamiento difuso del páncreas junto con áreas hipodensas.71 Los ganglios linfáticos peripancreáticos están presentes en algunas ocasiones y se visualizan tanto con la ECO como con la TC. La colangiopancreatografía retrógrada endoscópica puede demostrar el desplazamiento y la estenosis del conducto pancreático principal que suele confundirse con el tumor de páncreas.69 También se informó que la TBC pancreática se asocia con calcificación. A veces pueden observarse calcificaciones pequeńas.15 Las burbujas de gas en la TBC pancreática, que le otorgan apariencia enfisematosa, pueden representar un signo de sobreinfección.72 Se debe considerar en el diagnóstico diferencial la neoplasia pancreática, los abcesos y la pancreatitis crónica y el diagnóstico de tuberculosis pancreática debe realizarse mediante biopsia percutánea.
Hígado, vesícula y bazo
La TBC del hígado y del bazo es extremadamente frecuente en las autopsias de los pacientes con enfermedad diseminada; no obstante, no es habitual que se identifique en la presentación inicial.2 Cuando esto ocurre, suele encontrarse la diseminación miliar hepatoesplénica en asociación con la TBC miliar pulmonar. En esta forma de TBC se observa el compromiso hepatoesplénico micronodular, que se detecta como hepatoesplenomegalia moderada.4,16,18,26 Las lesiones individuales suelen encontrarse por debajo de la capacidad de resolución de las modalidades del diagnóstico por imágenes, pero en la TC pueden presentarse como pequeńos focos de baja densidad ampliamente distribuidos en el hígado (figura 17) y el bazo. La ecografía puede revelar el aumento de la ecogenicidad, patrón de “hígado brillante”. Estas características radiológicas no son específicas y se observan en diversas patologías benignas y malignas.
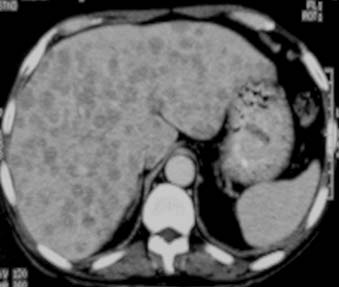
Figura 17
La forma macronodular (seudotumor o tuberculoma) es una manifestación rara de la TBC hepática o esplénica.18,73,74 Suele ocurrir sin compromiso manifiesto pulmonar o del tracto gastrointestinal, lo que sugiere una fuente de infección hematógena. Se puede visualizar como el agrandamiento difuso del hígado o del bazo, con contienen lesiones redondas múltiples de baja atenuación (15-50 UH) de 1 a 3 cm o como una masa única semejante a un tumor. La necrosis de estas lesiones puede conducir a la formación de un abceso. En etapas tempranas de la lesión, su apariencia típica es semejante a la del abceso con lesiones únicas o múltiples, de baja densidad tabicadas o “en panal” con márgenes irregulares mal definidos.56,74-76 Luego de la administración endovenosa de contraste se observa captación central mínima o escasa. Con su evolución, la región central se torna homogénea, pero se puede observar el relleno en la periferia.74 En la ECO detectamos que estas lesiones se presentan como lesiones redondas, tabicadas y ecogénicas (figuras 18a y 18b).42,25 No es habitual que los tabiques se visualicen con la TC (figura 18c), pero algunos autores demostraron que la apariencia en la TC y la ECO es similar.77 Con el tiempo la lesión se calcifica (en ocasiones con calcificación extensa).56,75,78
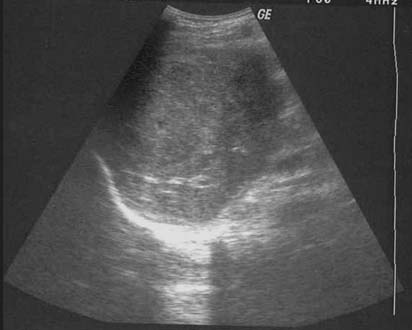
Figura 18a
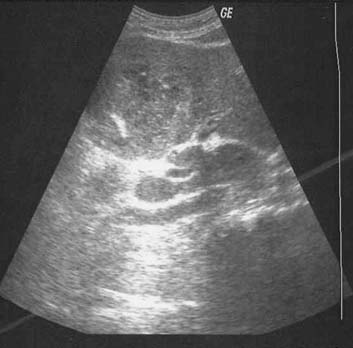
Figura 18b
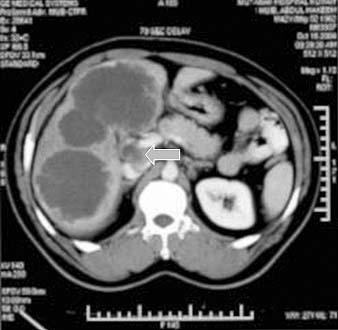
Figura 18c
En la RMN el tuberculoma solitario aparece hipointenso con márgenes hipointensos en las imágenes ponderadas en T1 e hiperintenso con márgenes menos intensos en las imágenes ponderadas en T2.79 Murata y col.80 informaron que en la RMN los tuberculomas hepáticos macronodulares se presentan como lesiones de baja intensidad en las imágenes ponderadas en T1 y T2, con relleno heterogéneo en las imágenes con contraste. Las lesiones esplénicas pueden ser el único signo de compromiso tuberculoso. En estos casos se debe considerar el linfoma en el diagnóstico diferencial.
HIV relacionado con la tuberculosis abdominal
En los pacientes con sida, el agente causal de la enfermedad por micobacterias es habitualmente Mycobacterium tuberculosis (MTB) y en ocasiones el complejo M. avium (CMA) es el agente responsable.54 La infección por MTB en los pacientes con sida produce imágenes radiográficas similares a las observadas en los pacientes sin sida, excepto en la enfermedad avanzada.54,67 El compromiso linfático es la característica más común en las personas con sida. En estos casos, los ganglios generalmente se localizan en la región mesentérica y en la peritoneal.81 La apariencia radiológica de la linfadenitis tuberculosa en los pacientes con sida o sin sida es similar. La mayoría presenta ganglios agrandados múltiples con realce periférico en la TC con contraste y centros de baja atenuación de tejido blando debido a la necrosis.82 No obstante, un cuarto de los sujetos con sida puede presentar ganglios agrandados de baja atenuación que no captan el contraste, un patrón que no se observa en los individuos sin sida.83 Esta posibilidad refleja la falta de reacción inflamatoria secundaria a la disfunción inmunológica.
La apariencia de los ganglios linfáticos abdominales es de gran utilidad para diferenciar la infección por MTB de la infección por el CMA,84-85 y estas diferencias podrían ser atribuidas a la respuesta del huésped al MTB y al CMA.
El CMA desencadena una menor respuesta tisular, con la formación del granuloma y caseificación, por lo que la necrosis (de baja atenuación) de los ganglios linfáticos es menor. La frecuencia de ganglios linfáticos con baja atenuación central es tan alta como del 93% en los pacientes con infección por MTB.84,85 La infección por CMA en los pacientes con sida produce linfadenopatías retroperitoneales y mesentéricas de gran tamańo, similares a las observadas en el linfoma y en el sarcoma de Kaposi metastásico.86,87
En la TBC del tracto GI, el sida puede comprometer el esófago con ulceración profunda, inflamación transmural y formación de fístulas y senos.88,89 En el estómago puede presentarse como una gran masa ulcerada en la pared gástrica con características benignas y malignas en las seriadas con bario del tracto GI superior y en la TC.90 Esto se manifiesta en la radiografía con bario como bario diluido debido al aumento de secreciones, dilatación difusa del intestino delgado con engrosamiento irregular de sus capas, habitualmente de distribución proximal que remedan la enfermedad de Whipple.91 El engrosamiento difuso de la pared yeyunal en la TC se informó en el 18% de los pacientes con infección por CMA.84 En estadios avanzados, la región ileocecal es el sitio que se compromete en forma primaria, las anormalidades colónicas son más extensas, el compromiso inflamatorio en la circunferencia del mesenterio circundante es mayor y aparecen las linfadenopatías de gran tamańo.54 También puede observarse la afección de sitios que no se comprometen con frecuencia, como el duodeno.67 El intestino grueso se encuentra afectado en el 9% de los casos sin la participación del intestino delgado.2 La enfermedad colónica puede presentarse con intususcepción, espasmo y rigidez de la lesión segmentaria. La estenosis, pequeńa o extensa, se manifiesta en los casos avanzados y debe diferenciarse de la colitis ulcerosa, enfermedad de Crohn, amebiasis, colitis isquémica, colitis seudomembranosa y cáncer de colon.92 La complicación de la TBC colónica es la perforación que produce peritonitis.92
La infección por CAM en los pacientes con sida provoca hepatomegalia importante en el 20% de los pacientes y esplenomegalia en el 14%.84 La infección por CAM también puede ser la causante de abcesos focales dentro del bazo y del hígado y en cualquier otro órgano macizo del abdomen.84 En el tipo miliar, la esplenomegalia se presenta en la ECO como el agrandamiento inespecífico y homogéneo del bazo. El compromiso esplénico macronodular se manifiesta en el 15% de los casos HIV positivos. La infección por MTB del páncreas en los pacientes con sida conduce a la formación de abcesos focales.84 La infección por CAM habitualmente produce un cuadro de pancreatitis crónica, que se visualiza en la pancreatografía como estrechamiento y dilatación de los conductos pancreáticos.93
La colangitis por CMA en los pacientes con sida provoca la estenosis ductal intrahepática y extrahepática y dilatación similar a la que se observa en la colangitis esclerosante primaria. Estas características son indistinguibles de las producidas por infecciones más comunes como la infección por citomegalovirus o por Criptosporidium.
El compromiso de la vesícula biliar es muy infrecuente y se puede visualizar el engrosamiento de la pared, tabiques ásperos e irregulares y linfadenopatía regional. El diagnóstico se realiza en mayor medida mediante la histopatología más que por los hallazgos radiológicos.
Los autores no manifiestan “conflictos de interés”.
BIBLIOGRAFÍA
-
Carrera GF, Young S, Lewicki AM. Intestinal tuberculosis. Gastrointest Radiol
1976; 1:147-55.
-
Thoeni RF, Margulis AR. Gastrointestinal tuberculosis. Semin Roentgenol 1979;
14:283-94.
-
Manohar A, Simjee AE, Haffejee AA, Pettengell KE. Symptoms and investigative
findings in 145 patients with tuberculous peritonitis diagnosed by peritoneoscopy
and biopsy over a five year period. Gut 1990; 31:1130-2.
-
Bhansali SK. Abdominal tuberculosis. Experiences with 300 cases. Am J
Gastroenterol 1977; 67:324-37.
-
Al-Hadeedi S, Walia HS, Al-Sayer HM. Abdominal tuberculosis. Can J Surg
1990; 33:233-7.
-
Jakubowski A, Elwood RK, Enarson DA. Clinical features of abdominal
tuberculosis. J Infect Dis 1988; 158:687-92.
-
Crowley JJ, Ramji FG, Amundson GM. Genital tract tuberculosis with peritoneal
involvement: MR appearance. Abdom Imaging 1997; 22:445-7.
-
Yapar EG, Ekici E, Karasahin E, Gokmen O. Sonographic features of tuberculous
peritonitis with female genital tract tuberculosis. Ultrasound Obstet Gynecol 1995;
6:121-5.
-
Epstein BM, Mann JH. CT of abdominal tuberculosis. AJR Am J Roentgenol
1982; 139:861-6.
-
Hanson RD, Hunter TB. Tuberculous peritonitis: CT appearance. AJR Am J
Roentgenol 1985; 144:931-2.
-
Batra A, Gulati MS, Sarma D, Paul SB. Sonographic appearances in abdominal
tuberculosis. J Clin Ultrasound 2000; 28:233-45.
-
Leder RA, Low VH. Tuberculosis of the abdomen. Radiol Clin North Am 1995;
33:691-705.
-
Lee DH, Lim JH, Ko YT, Yoon Y. Sonographic findings in tuberculous peritonitis
of wet-ascitic type. Clin Radiol 1991; 44:306-10.
-
Jadvar H, Mindelzun RE, Olcott EW, Levitt DB. Still the great mimicker:
abdominal tuberculosis. AJR Am J Roentgenol 1997; 168:1455-60.
-
Denton T, Hossain J. A radiological study of abdominal tuberculosis in a Saudi
population, with special reference to ultrasound and computed tomography. Clin
Radiol 1993; 47:409-14.
-
Ramaiya LI, Walter DF. Sonographic features of tuberculous peritonitis. Abdom
Imaging 1993; 18:23-6.
-
Kedar RP, Shah PP, Shivde RS, Malde HM. Sonographic findings in
gastrointestinal and peritoneal tuberculosis. Clin Radiol 1994; 49:24-9.
-
Hulnick DH, Megibow AJ, Naidich DP, Hilton S, Cho KC, Balthazar EJ. Abdominal
tuberculosis: CT evaluation. Radiology 1985; 157:199-204.
-
Jorge AD. Peritoneal tuberculosis. Endoscopy 1984; 16:10-2.
-
Sinan T, Sheikh M, Ramadan S, Sahwney S, Behbehani A. CT features in
abdominal tuberculosis: 20 years experience. BMC Med Imaging 2002; 2:3
-
Akhan O, Demirkazik FB, Demirkazik A, Gulekon N, Eryilmaz M, Unsal M, et al.
Tuberculous peritonitis: ultrasonic diagnosis. J Clin Ultrasound 1990; 18:711-4.
-
Lundstedt C, Nyman R, Brismar J, Hugosson C, Kagevi I. Imaging of
tuberculosis. II. Abdominal manifestations in 112 patients. Acta Radiol 1996;
37:489-95.
-
Zirinsky K, Auh YH, Kneeland JB, Rubenstein WA, Kazam E. Computed
tomography, sonography, and MR imaging of abdominal tuberculosis. J Comput
Assist Tomogr 1985; 9:961-3.
-
Demirkazik FB, Akhan O, Ozmen MN, Akata D. US and CT findings in the
diagnosis of tuberculous peritonitis. Acta Radiol 1996; 37:517-20.
-
Sheikh M, Abu-Zidan F, al-Hilaly M, Behbehani A. Abdominal tuberculosis:
comparison of sonography and computed tomography. J Clin Ultrasound 1995;
23:413-7.
-
Dahlene DH Jr, Stanley RJ, Koehler RE, Shin MS, Tishler JM. Abdominal
tuberculosis: CT findings. J Comput Assist Tomogr 1984; 8:443-5.
-
Jain R, Sawhney S, Bhargava DK, Berry M. Diagnosis of abdominal
tuberculosis: sonographic findings in patients with early disease. AJR Am J
Roentgenol 1995; 165:1391-5.
-
Prasad S, Patankar T. Computed tomography demonstration of a fat-fluid level
in tuberculous chylous ascites. Australas Radiol 1999; 43:542-3.
-
Bankier AA, Fleischmann D, Wiesmayr MN, Putz D, Kontrus M, Hubsch P, et al.
Update: abdominal tuberculosis--unusual findings on CT. Clin Radiol 1995; 50:223-
8.
-
Rodriguez E, Pombo F. Peritoneal tuberculosis versus peritoneal
carcinomatosis: distinction based on CT findings. J Comput Assist Tomogr 1996;
20:269-72.
-
Ozkan K, Gurses N, Gurses N. Ultrasonic appearance of tuberculous peritonitis.
J Clin Ultrasound 1987; 15:350-2.
-
Wu CC, Chow KS, Lu TN, Huang FT. Sonographic features of turberculous
omental cakes in peritoneal tuberculosis. J Clin Ultrasound 1988; 16:195-8.
-
Ha HK, Jung JI, Lee MS, Choi BG, Lee MG, Kim YH, et al. CT differentiation of
tuberculous peritonitis and peritoneal carcinomatosis. AJR Am J Roentgenol 1996;
167:743-8.
-
Akhan O, Kalyoncu F, Ozmen MN, Demirkazik FB, Cekirge HS, Sahin A, et al.
Peritoneal mesothelioma: sonographic findings in nine cases. Abdom Imaging 1993;
18:280-2.
-
Arai K, Makino H, Morioka T, Yagi H, Minabe K, Takeyama S, et al.
Enhancement of ascites on MRI following intravenous administration of Gd-DTPA. J
Comput Assist Tomogr 1993; 17:617-22.
-
Skendros P, Kamaria F, Kontopoulos V, Tsitouridis I, Sidiropoulos L. Intradural,
eextramedullary tuberculoma of the spinal cord as a complication of tuberculous
meningitis. Infection 2003; 31:115-7.
-
Rossner R. [50 years of x-ray mass screening]. Rontgenpraxis 1990; 43:376-9.
-
Kinoshita T, Yashiro N, Yoshigi J, Ihara N, Fukuma E, Narita M. Inflammatory
intramammary lymph node mimicking the malignant lesion in dynamic MRI: a case
report. Clin Imaging 2002; 26:258-62.
-
Gasparetto EL, Tazoniero P, de Carvalho Neto A. Disseminated tuberculosis in a
pregnant woman presenting with numerous brain tuberculomas: case report. Arq
Neuropsiquiatr 2003; 61:855-8.
-
Ebert DL, Olivier KN. Nontuberculous mycobacteria in the setting of cystic
fibrosis. Clin Chest Med 2002; 23:655-63.
-
Aggarwal D, Suri A, Mahapatra AK. Orbital tuberculosis with abscess. J
Neuroophthalmol 2002; 22:208-10.
-
Sheikh M, Moosa I, Hussein FM, Qurttom MA, Behbehani AI. Ultrasonographic
diagnosis in abdominal tuberculosis. Australas Radiol 1999; 43:175-9.
-
Da Costa H, Adatrao V, Merchant S. A radiocolloid study of the reticulo-
endothelial system in tropical diseases. Nuklearmedizin 1979; 18:130-2.
-
Neufang KF, Beyer D. [Conventional diagnosis of lymphadenopathies--value of
conventional roentgen examination and supplementation of other imaging
technics]. Rontgenblatter 1983; 36:30-44.
-
Stanley JH, Yantis PL, Marsh WH. Periportal tuberculous adenitis: a rare cause
of obstructive jaundice. Gastrointest Radiol 1984; 9:227-9.
-
Alvarez SZ, Carpio R. Hepatobiliary tuberculosis. Dig Dis Sci 1983; 28:193-200.
-
Caroli-Bosc FX, Conio M, Maes B, Chevallier P, Hastier P, Delmont JP.
Abdominal tuberculosis involving hepatic hilar lymph nodes. A cause of portal vein
thrombosis and portal hypertension. J Clin Gastroenterol 1997; 25:541-3.
-
Reichard AA, Lobato MN, Roberts CA, Bazerman LB, Hammett TM. Assessment
of tuberculosis screening and management practices of large jail systems. Public
Health Rep 2003; 118:500-7.
-
Pombo F, Rodriguez E, Mato J, Perez-Fontan J, Rivera E, Valvuena L. Patterns
of contrast enhancement of tuberculous lymph nodes demonstrated by computed
tomography. Clin Radiol 1992; 46:13-7.
-
Moon WK, Im JG, Yu IK, Lee SK, Yeon KM, Han MC. Mediastinal tuberculous
lymphadenitis: MR imaging appearance with clinicopathologic correlation. AJR Am J
Roentgenol 1996; 166:21-5.
-
Kim SY, Kim MJ, Chung JJ, Lee JT, Yoo HS. Abdominal tuberculous
lymphadenopathy: MR imaging findings. Abdom Imaging 2000; 25:627-32.
-
Vidal ML, del Castillo F, Arroba ML, Borque C, Garcia Hortelano J. [Abdominal
tuberculosis in children. A review apropos of 13 cases]. An Esp Pediatr 1986;
24:227-31.
-
Yang ZG, Min PQ, Sone S, He ZY, Liao ZY, Zhou XP, et al. Tuberculosis versus
lymphomas in the abdominal lymph nodes: evaluation with contrast-enhanced CT.
AJR Am J Roentgenol 1999; 172:619-23.
-
Balthazar EJ, Gordon R, Hulnick D. Ileocecal tuberculosis: CT and radiologic
evaluation. AJR Am J Roentgenol 1990; 154:499-503.
-
Brown JH, Berman JJ, Blickman JG, Chew FS. Primary ileocecal tuberculosis.
AJR Am J Roentgenol 1993; 160:278
-
Denath FM. Abdominal tuberculosis in children: CT findings. Gastrointest Radiol
1990; 15:303-6.
-
Suri R, Gupta S, Gupta SK, Singh K, Suri S. Ultrasound guided fine needle
aspiration cytology in abdominal tuberculosis. Br J Radiol 1998; 71:723-7.
-
Makanjuola D. Is it Crohn's disease or intestinal tuberculosis? CT analysis. Eur J
Radiol 1998; 28:55-61.
-
Tandon HD, Prakash A. Pathology of intestinal tuberculosis and its distinction
from Crohn’s disease. Gut 1972; 13:260-9.
-
McNamara M, Williams CE, Brown TS, Gopichandran TD. Tuberculosis affecting
the oesophagus. Clin Radiol 1987; 38:419-22.
-
Tornieporth N, Lorenz R, Gain T, Rosch T, Classen M. An unusual case of active
tuberculosis of the oesophagus in an adult. Endoscopy 1991; 23:294-6.
-
Okoro EO, Komolafe OF. Gastric tuberculosis: unusual presentations in two
patients. Clin Radiol 1999; 54:257-9.
-
Fukuya T, Yoshimitsu K, Kitagawa S, Masuda K, Ueyama T, Haraguchi Y. Single
tuberculous stricture in the jejunum: report of 2 cases. Gastrointest Radiol 1989;
14:300-4.
-
Healy JC, Gorman S, Kumar PJ. Case report: tuberculous colitis mimicking
Crohn’s disease. Clin Radiol 1992; 46:131-2.
-
Nakano H, Jaramillo E, Watanabe M, Miyachi I, Takahama K, Itoh M. Intestinal
tuberculosis: findings on double-contrast barium enema. Gastrointest Radiol 1992;
17:108-14.
-
Downey DB, Nakielny RA. Aphthoid ulcers in colonic tuberculosis. Br J Radiol
1985; 58:561-2.
-
Bargallo N, Nicolau C, Luburich P, Ayuso C, Cardenal C, Gimeno F. Intestinal
tuberculosis in AIDS. Gastrointest Radiol 1992; 17:115-8.
-
Lee DH, Ko YT, Yoon Y, Lim JH. Sonographic findings of intestinal tuberculosis.
J Ultrasound Med 1993; 12:537-40.
-
Fischer G, Spengler U, Neubrand M, Sauerbruch T. Isolated tuberculosis of the
pancreas masquerading as a pancreatic mass. Am J Gastroenterol 1995; 90:2227-
30.
-
Desai DC, Swaroop VS, Mohandas KM, Borges A, Dhir V, Nagral A, et al.
Tuberculosis of the pancreas: report of three cases. Am J Gastroenterol 1991;
86:761-3.
-
Desai SR, Bhanthunmavin K, Hollands M. Primary pancreatic tuberculosis:
presentation and diagnosis. Aust N Z J Surg 2000; 70:141-3.
-
Morris DL, Wilkinson LS, al Mokhtar N. Case report: emphysematous
tuberculous pancreatitis diagnosis by ultrasound and computed tomography. Clin
Radiol 1993; 48:286-7.
-
Choi BI, Im JG, Han MC, Lee HS. Hepatosplenic tuberculosis with
hypersplenism: CT evaluation. Gastrointest Radiol 1989; 14:265-7.
-
Kapoor R, Jain AK, Chaturvedi U, Saha MM. Ultrasound detection of
tuberculomas of the spleen. Clin Radiol 1991; 43:128-9.
-
Buxi TB, Vohra RB, Sujatha Y, Chawla D, Byotra SP, Gupta PS, et al. CT
appearances in macronodular hepatosplenic tuberculosis: a review with five
additional new cases. Comput Med Imaging Graph 1992; 16:381-7.
-
Wilde CC, Kueh YK. Case report: Tuberculous hepatic and splenic abscess. Clin
Radiol 1991; 43:215-6.
-
Blangy S, Cornud F, Sibert A, Vissuzaine C, Saraux JL, Benacerraf R. Hepatitis
tuberculosis presenting as tumoral disease on ultrasonography. Gastrointest Radiol
1988; 13:52-4.
-
Malde HM, Chadha D. The "cluster" sign in macronodular hepatic tuberculosis:
CT features. J Comput Assist Tomogr 1993; 17:159-61.
-
Kawamori Y, Matsui O, Kitagawa K, Kadoya M, Takashima T, Yamahana T.
Macronodular tuberculoma of the liver: CT and MR findings. AJR Am J Roentgenol
1992; 158:311-3.
-
Murata Y, Yamada I, Sumiya Y, Shichijo Y, Suzuki Y. Abdominal macronodular
tuberculomas: MR findings. J Comput Assist Tomogr 1996; 20:643-6.
-
Schanaider A, Madi K. Intra-abdominal tuberculosis in acquired
immunodeficiency syndrome. Diagnosis and management. Int Surg 1995; 80:147-
51.
-
Kaplan MM, Boice JD Jr, Ames DB, Rosenstein M. Thyroid, parathyroid, and
salivary gland evaluations in patients exposed to multiple fluoroscopic examinations
during tuberculosis therapy: a pilot study. J Clin Endocrinol Metab 1988; 66:376-
82.
-
Greenberg AK, Knapp J, Rom WN, Addrizzo-Harris DJ. Clinical presentation of
pulmonary mycetoma in HIV-infected patients. Chest 2002; 122:886-92.
-
Radin DR. Intraabdominal Mycobacterium tuberculosis vs Mycobacterium
avium-intracellulare infections in patients with AIDS: distinction based on CT
findings. AJR Am J Roentgenol 1991; 156:487-91.
-
Nyberg DA, Federle MP, Jeffrey RB, Bottles K, Wofsy CB. Abdominal CT findings
of disseminated Mycobacterium avium-intracellulare in AIDS. AJR Am J Roentgenol
1985; 145:297-9.
-
Prasad RS, Fraser MH, Urquhart GD, McLean AN. Rupture of tuberculous spinal
abscess resulting in tuberculous empyema and chylothorax. Spinal Cord 2003;
41:410-2.
-
Von Kummer R, Storch B, Rauch H, Krause KH. [A case of multiple intracranial
tuberculoma followed by serial computerized tomography (author's transl)].
Nervenarzt 1981; 52:344-7.
-
De Silva R, Stoopack PM, Raufman JP. Esophageal fistulas associated with
mycobacterial infection in patients at risk for AIDS. Radiology 1990; 175:449-53.
-
Goodman P, Pinero SS, Rance RM, Mansell PW, Uribe-Botero G. Mycobacterial
esophagitis in AIDS. Gastrointest Radiol 1989; 14:103-5.
-
Brody JM, Miller DK, Zeman RK, Klappenbach RS, Jaffe MH, Clark LR, et al.
Gastric tuberculosis: a manifestation of acquired immunodeficiency syndrome.
Radiology 1986; 159:347-8.
-
Vincent ME, Robbins AH. Mycobacterium avium-intracellulare complex enteritis:
pseudo-Whipple disease in AIDS. AJR Am J Roentgenol 1985; 144:921-2.
-
Gupta NM, Motup T, Joshi K. Isolated colonic tuberculous perforation as a rare
cause of peritonitis: report of a case. Surg Today 1999; 29:273-5.
-
Farman J, Brunetti J, Baer JW, Freiman H, Comer GM, Scholz FJ, et al. AIDS-
related cholangiopancreatographic changes. Abdom Imaging 1994; 19:417-22.
|






