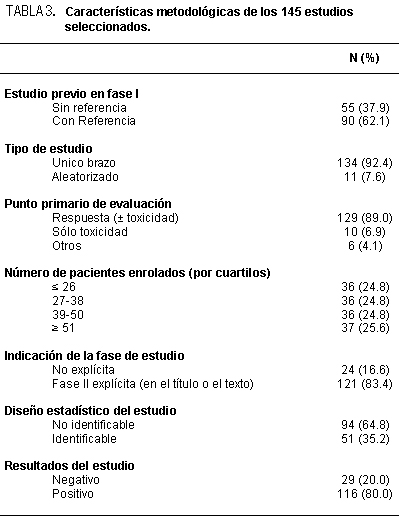
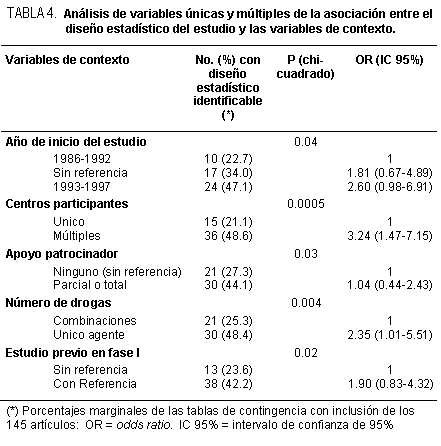
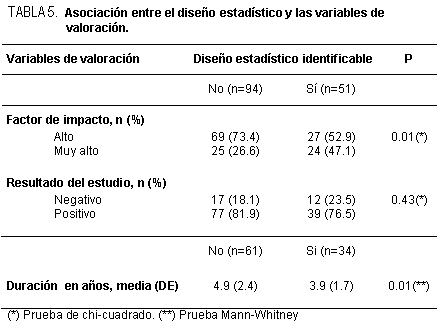
En los ensayos clínicos donde se comparan dos formas de manejo de los
integrantes de la investigación, se pueden describir varias maneras de expresar los
resultados. Desde luego, si éstos son dicotómicos y se ordenan los valores numéricos en
una tabla de 2 x 2, podríamos comparar la distribución de los valores obtenidos con la
distribución teórica bajo la hipótesis nula de que no hay diferencias de los grupos en
comparación: chi cuadrado, entonces. Ello nos dará en forma general la información de
cuán probable es que los resultados que se han volcado en la tabla tengan una distribución
atribuible o explicable por el azar. Si no se cumplen los requisitos que exige esta prueba de
hipótesis, se pueden analizar los datos empleando la prueba de Fisher-Irwin de
probabilidades exactas. Igualmente, se puede manejar como la comparación de dos
proporciones por una aproximación a «Z» así como analizarlos de acuerdo con sus
intervalos de confianza respectivos. Como sea, se obtendrá "p", es decir el error alfa. Hasta
aquí no se tendrá más información. Sin embargo, para cada grupo tenemos a disposición el
total de casos y la parte que tuvo el resultado en estudio. De tal manera, veremos que la
información logra ir bastante más lejos que lo indicado hasta este instante. Si consideramos
los dos grupos sugeridos, sometidos a tratamiento experimental y control, esperando un
resultado determinado, tendríamos:
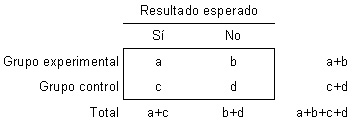
Asignando valores para uso posterior, supongamos que:
a = 12; b = 104; c = 28; d = 101.
El riesgo corresponde a la probabilidad de ocurrencia de un suceso (generalmente no
deseado). De este modo, si del grupo experimental se espera que tenga con menor
frecuencia un resultado adverso («sí», en la tabla), es decir que el tratamiento experimental
tenga efecto benéfico, la presentación de la información puede ser como a continuación:
Riesgo absoluto (RA) en el grupo control = c/(c+d) = 28/129 = 0.217
Riesgo absoluto (RA) en el grupo experimental = a/(a+b) = 12/116 = 0.103
Riesgo relativo (RR) = (RA del grupo experimental)/(RA del grupo control)
= 0.103 / 0.217 = 0.48
IC 95% del RR, 0.25 – 0.89 (por Statcalc de EpiInfo)
El riesgo relativo (RR) seńala cuánto más probable o menos probable es el efecto o
resultado en el grupo experimental que en el grupo control. Al valor obtenido se puede
agregar su intervalo de confianza, generalmente del 95%, que nos indicará las cifras entre
las cuales se encontraría, con un 95% de probabilidad, el RR del universo del que proceden
los casos estudiados. Para que el RR encontrado en la investigación pueda considerarse de
interés, su IC no debe incluir el valor «1», puesto que esa cifra indica riesgos absolutos
iguales.
La llamada «reducción del riesgo absoluto» (RRA) es simplemente la disminución del riesgo
observada del grupo control al grupo experimental y corresponde entonces a su diferencia
RA del grupo control - RA del grupo experimental = RRA
0.217 – 0.103 = 0.114
21.7% - 10.3% = 11.4%
IC 95% de la RRA, 0.0239 – 0.2041 (2.39% - 20.41%) (por MedCalc)
Otra expresión de interés es la «reducción relativa del riesgo» (RRR) (no se equivoque, esta
no es «reducción del riesgo relativo») que es la proporción que representa la reducción del
riesgo absoluto (RRA) respecto del riesgo basal, que sería el del grupo control. Entonces
(RRA/RA del grupo control) x 100 = RRR
Si tenemos el valor de la reducción del riesgo absoluto y ésta es de, como se estableció en
el ejemplo, 0.114 o 11.4%, podemos interpretarla en el sentido que de cada 100 sujetos
tratados con el esquema experimental 11.4 de ellos obtendrán el beneficio (adicional) que
representa el nuevo tratamiento. Ahora, 1 de ellos obtendrá el beneficio adicional por cada
100/11.4 = 8.77 tratados (la cifra se redondea al entero superior, de modo que quedaría en
9 casos). Esto representa el llamado «número necesario a tratar» (NNT) para obtener 1
caso adicional beneficiado. Habitualmente se le expresa como el valor inverso de la
reducción del riesgo absoluto. Para el ejemplo planteado, en que el tratamiento
experimental reduce el riesgo de un suceso indeseado, 9 pacientes deben ser tratados en el
grupo experimental para evitar un episodio adicional no deseado.
Conviene recordar que la expresión de un resultado sólo como RR plantea un problema, ya
que por tratarse de una relación entre dos riesgos absolutos, un mismo valor del RR puede
representar riesgos absolutos totalmente diferentes. Así, un RR de 2 puede ser el resultado
de 0.40 / 0.20 o 0.05 / 0.025 , etc. En ambos casos la RRR es 50% , sin embargo NNT en la
primera situación es de 5 casos y en el segundo es 40 casos, lo que con toda seguridad
afectará las decisiones clínicas.
El intervalo de confianza del NNT no parece ser un asunto definitivamente resuelto, pero se
le puede estimar utilizando los valores inversos de los extremos del intervalo de confianza
de la RRA : 1 / 0.0239 o 100 / 2.39 = 41.8 ,es decir 42 casos a 1 / 0.2041 o 100 / 20.41 =
4.89, es decir 5 casos. Entonces, para la situación propuesta, NNT es 9 pacientes con un IC
del 95% de 5 a 42.
Cuando el IC de la RRA pasa por cero, es decir cuando los riesgos absolutos no se pueden
considerar significativamente diferentes, el IC 95% del NNT presentará un problema de
interpretación. Supongamos una RRA de 10% (0.10) con IC 95% de -5% a +25%. En tal
caso NNT sería 10 con extremos del IC de –20 a + 4. Lo que inmediatamente llamaría la
atención es que el valor del NNT , 10, no parece estar contenido en su intervalo de
confianza. Esto no sería tal si vemos lo siguiente:
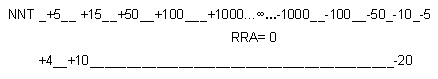
Una lectura de este IC 95% sería, por ejemplo, que se requiere tratar 10 pacientes para
obtener un caso adicional con el beneficio deseado variando desde 4 pacientes para obtener
tal beneficio en uno adicional hasta 20 para reducir en un caso los favorecidos, comparado
con el tratamiento de contraste.
Puesto que los casos de un grupo son heterogéneos en su nivel de riesgo, se podría
hacer un intento (un tanto difícil) de caracterizar el paciente individual en cuanto a su
probabilidad específica (expresada como una fracción) de un evento respecto al valor medio
con el que se obtuvo el NNT del grupo. Una vez estimada esta fracción para un caso
determinado, se divide NNT por ella y se obtendrá el NNT para ése tipo de paciente.
Cuando el tratamiento experimental aumenta la probabilidad de un suceso favorable
los índices se modifican un tanto y así tendremos el aumento relativo del beneficio (ARB) en
que se comparará el incremento de frecuencia de sucesos favorables con la frecuencia basal
dada por el grupo control. Así, si en éste último la frecuencia es 30% y en el grupo
experimental 40%, el incremento de 10% se expresará en relación al 30% del grupo control
y obtendremos
(10% / 30%) x 100 = 33.33%
Este valor es en definitiva el ARB. El aumento absoluto del beneficio (AAB) se expresa como
la diferencia simple entre los dos grupos respecto a la frecuencia de sucesos beneficiosos
considerados. En el ejemplo, AAB sería de 10%. Su valor inverso corresponde al NNT, que
aquí sería 100/10 = 10 es decir, 10 casos deben recibir el tratamiento experimental para
obtener la ocurrencia de un suceso favorable adicional, comparado con el grupo control.
Si el tratamiento experimental causa cierto dańo, es decir cuando aumenta la
probabilidad de un suceso no deseado, comparado con el tratamiento control, nuevamente
la situación cambia algo en el análisis de los resultados. Así, el aumento relativo del riesgo
(ARR) se calcularía del mismo modo que el ARB, sólo que en vez de referirse al aumento del
beneficio lo hace respecto al aumento del riesgo. Nuevamente tenemos aquí la opción de
medir el cambio del riesgo absoluto, en este caso aumento del mismo (ARA), lo que
corresponde a la diferencia de riesgo en ambos grupos de tratamiento.
Como en los casos anteriores, el valor inverso de ARA proporcionará el número
de pacientes con tratamiento experimental necesarios para generar un caso adicional de
dańo comparado con la situación del grupo control (NND).
El valor de NNT puede ser obtenido también en los casos en que se dispone sólo del
riesgo relativo (RR) o de la razón de disparidad (OR):
A partir del RR.
Si el RR es menor que 1:
NNT = 1 / ( 1 – RR ) x Fc
Fc= Frecuencia esperada en los controles.
Con los datos de la tabla vemos que el grupo experimental tiene menor riesgo que los
controles 0.103 vs. 0.217, lo que indicaría que el tratamiento nuevo es «protector» y el RR
es inferior a 1, es decir, 0,4746. Entonces,
NNT = 1 / ( 1 – 0.4746 ) x 0.217 = 8.77
y se redondea a 9, cifra que coincide con la encontrada previamente.
Si el RR es mayor que 1:
NNT = 1 / ( RR – 1) x Fc.
El mismo ejemplo anterior Ud. podría verlo desde el punto de vista inverso, es decir que el
riesgo en los controles es mayor que en el grupo experimental y el RR sería 0.217 / 0.103 =
2.10. Empleando la fórmula propuesta para el caso en que RR es mayor que 1:
NNT = 1 / (2.1068 – 1 ) x 0.103 = 8,77
que se redondea a 9.
Como se puede ver, el referente es el grupo experimental, que aquí pasaría a ser «control»
respecto del cual se informará del resultado del otro grupo.
Lo que cambia, de una a otra forma de cálculo, es la lectura del resultado. En el primer caso
«se requiere tratar a 9 pacientes con el tratamiento experimental ("protector") para evitar
el riesgo en un paciente adicional respecto del grupo (tratamiento) control". En el segundo
caso "se requiere tratar a 9 pacientes para obtener un caso adicional padeciendo el riesgo
asociado al tratamiento control".
Cuando se dispone de OR, se puede obtener NNT de la siguiente manera:
Si OR es menor que 1:
NNT = 1 - [ p0 ( 1 – OR ) ] / (1 - p0) p0 ( 1 - OR)
Si OR es mayor que 1 :
NNT = 1 + [ p0 ( OR - 1 ) ] / (1 - p0) p0 ( OR - 1 )
p0 = Frecuencia esperada en el paciente (exposición en los controles).
En un ejemplo:

p0 = Exposición en controles = 150/230 = 0.65
OR= 2.67
NNT = 1 + [0.65 (2.67 – 1)] / (1 – 0.65) 0.65 (2.67 – 1) = 5.48
cifra que se aproxima a 6. Se lee así: Por cada 6 expuestos, se agrega un "caso" adicional.
Umbral de NNT. Si bien en muchas oportunidades, conociendo el resultado de NNT
se podrá intuitivamente considerar que es una cifra de interés, en otros casos se requerirá
establecer un umbral, que si se trata de obtener un beneficio por ejemplo, permitirá decir
que si NNT está por debajo de él es claramente conveniente, pero si está por encima muy
probablemente ya no lo es. Dos formas de estimación que se han propuesto, trabajan con el
aspecto económico una de ellas y con el aspecto clínico la otra.
NNT calculado por programas de computación. Mencionaremos que variados
programas ofrecen esta función pero, como siempre, si el usuario no está familiarizado con
las bases bioestadísticas que subyacen, es muy probable que pueda equivocarse en la
introducción de la información o en la interpretación de los resultados.
BIBLIOGRAFÍA
-
-Sackett DL, Richardson WS, Rosenberg W, Haynes RB. Evidence based medicine. How
to practice and teach EBM.London: Churchill Livingstone, 1997: 136-141 , 168-170.
-
-Altman DG.: Confidence intervals for the number needed to treat. BMJ , 1998; 317:
1309- 1312.
-
-Altman DG, Andersen PK: Calculating the number needed to treat for trials where the
outcome is time to an event. BMJ,1999;319:1492-1495.
-
-Smeeth L., Haines A., Ebrahim S.: Numbers needed to treat derived from meta-
analyses. BMJ, 1999; 318:1548 – 1551.