Las extremidades superiores e inferiores son una compilación de
compartimientos que contienen músculos, nervios y vasos envueltos por fascias
o tensas. El síndrome compartimental se asocia con un aumento en la presión
dentro de estos envoltorios tensos, y pone en riesgo la viabilidad de las
estructuras incluidas.1,2
Fisiopatología
Isquemia:
La lesión de una arteria dominante de una extremidad
causa la hinchazón o el agrandamiento posisquémico del tejido debido a la
trasudación de líquido a través de los capilares de las membranas
basales.3,4 Esto conduce a la compresión de los capilares y a la
isquemia tisular.
Reperfusión: Resulta en la producción de radicales libres que potencian
la agresión a las membranas celulares ya dańadas y promueven la agregación
plaquetaria y la coagulación de microvasos.5,6 La coagulación
intravascular crea mayor anoxia y el círculo vicioso continúa.
Oclusión venosa: Aunque es poco común, la lesión y la trombosis
ulterior de venas mayores de las extremidades pueden inducir anoxia por sí
mismas debido a ectasia venosa, agrandamiento masivo agudo y compresión
capilar.7,8 Es más común luego de la ligadura de las venas
dańadas en lesiones combinadas arteriales y venosas.
Síntomas y signos
El pilar diagnóstico del síndrome compartimental es sospecharlo y
diagnosticarlo tempranamente. Las seis P (pain [dolor], presión,
parestesias, parálisis, ausencia de pulsos y palidez) se utilizan como regla
mnemotécnica fácil para el diagnóstico clínico.9
El dolor es probablemente el síntoma más sensible porque es informado en
forma invariable por todos los pacientes conscientes con una extremidad
lesionada. Es el dolor desproporcionado con respecto a la lesión existente lo que
alertará al facultativo experimentado. La presión se refiere a la sensación de
tensión durante la palpación de la extremidad comprometida. Es probable que las
parestesias sean los primeros síntomas en aparecer debido a que los nervios son
muy sensibles a la lesión. La parálisis es causada tanto por compresión nerviosa
prolongada como por dańo muscular irreversible. Es casi siempre una
manifestación tardía y una indicación de que el diagnóstico se retrasó. La
ausencia de pulsos es en realidad otro síntoma que nunca debería estar presente
si el diagnóstico del síndrome compartimental fue realizado a tiempo. El mismo
principio previamente descrito se aplica a la palidez. Así, en ubicación distal a los
compartimientos involucrados, la perfusión se conserva debido al flujo sanguíneo
inalterado a través de las arterias mayores, el color de la piel debería ser normal
(rosado y no pálido).
Medición de las presiones y pruebas de laboratorio
Monitoreo de la presión intracompartimental
La presión en el compartimiento puede medirse directamente mediante la
introducción de una aguja conectada a un transductor de presión. El dispositivo
más usado es el sistema de monitoreo de presión intracompartimental de
Stryker. Es importante medir todos los compartimientos en el área de la
extremidad de interés. Las presiones menores a 20 mm Hg son aceptables,
mientras que las mayores a 30 mm Hg generalmente indican la presencia de un
síndrome compartimental. Los valores comprendidos entre ambos pertenecen a
la "zona gris".1,2,10 No existe un umbral absoluto de presión que
esté confiablemente asociado con el síndrome compartimental. Un examen
clínico minucioso es más importante. La combinación de hallazgos clínicos y las
mediciones de presión establecerán el diagnóstico. Más que la presión
intracompartimental absoluta, se ha sugerido la presión de perfusión como
indicador más fiable del síndrome compartimental: es la diferencia entre la
presión arterial media y la presión intracompartimental, también conocida como
delta p (ΔP). Luego de fractura tibial se mostró que si la ΔP
permanece satisfactoria, los pacientes con presiones intramusculares no tienen
mayor incidencia de síndrome compartimental que aquellos con presiones
bajas.11
Creatina fosfoquinasa sérica y mioglobina
La creatina fosfoquinasa sérica (CPK) ha sido utilizada como marcador del
síndrome compartimental.12,13 Esta enzima indica necrosis
muscular.13,14 Así, las elevaciones más allá de los niveles
normales equivalen a dańo permanente de fibras musculares. La CPK no es
apropiada para la detección temprana, pero sus tendencias son útiles para
monitorear la progresión de síndromes equívocos o de compartimientos
recientemente descomprimidos. Cuando el diagnóstico del síndrome no es claro
la determinación de la CPK puede utilizarse como herramienta adicional para
tomar una decisión. Los niveles de más de 520 U son anormales y los superiores
a 5 000 U se asocian con dańo muscular importante y colocan al paciente con
riesgo elevado de falla renal. En pacientes críticamente lesionados, el pico de
CPK se produce dentro de las 96 horas y un nuevo aumento o persistencia de
valores elevados luego de su pico pueden indicar nuevo dańo muscular o dańo
muscular en curso. De forma similar, luego de la descompresión, los niveles de
CPK deberían mostrar tendencia al descenso.
La mioglobinuria es también otro marcador de lisis de células
musculares.15,16 Puede ser diagnosticada en forma errónea y
confundida con hematuria. Una prueba urinaria de benzidina para sangre oculta
en ausencia de eritrocitos es la clave para el diagnóstico.
Otros hallazgos de laboratorio, como anemia, hiperkalemia, hipocalcemia,
hiperfosfatemia, trombocitopenia, uremia y acidosis metabólica sirven sólo para
indicar la carencia de tratamiento temprano de síndrome
compartimental.15,16
Oximetría de pulso y espectroscopia cercana al infrarrojo
Se propuso la oximetría de pulso como ayuda para el monitoreo del
aumento de las presiones intracompartimentales pero no resultó confiable. La
espectroscopia cercana al infrarrojo es un método nuevo que puede evaluar el
nivel de oxihemoglobina muscular.17,18 La saturación de la
oxihemoglobina normal del músculo es aproximadamente 85%. Los valores
menores a 60% se correlacionan con el síndrome compartimental. La limitada
investigación realizada hasta la fecha con esta técnica no permite obtener
conclusiones seguras sobre su uso.
Tratamiento
El estándar actual de cuidado obliga a la descompresión quirúrgica para los
síndromes compartimentales establecidos.1,2,13,18,19-22 La
fasciotomía realizada a lo largo del eje longitudinal del compartimiento entero
libera la presión y restaura la microperfusión. Los elementos más importantes de
una fasciotomía exitosa son las incisiones adecuadas de la piel y de las fascias.
Extremidades inferiores
Para completar una fasciotomía de la pierna en cuatro compartimientos se
utiliza una incisión lateral y una medial (Figuras 1 y 2). En forma alternativa, una
única incisión lateral puede usarse para descomprimir los cuatro
compartimientos23 (Figura 3). En el muslo se utiliza una incisión
lateral y una medial (Figura 4), y en las circunstancias infrecuentes de síndrome
compartimental del pie. Una incisión cutánea lateral semicircular se emplea para
los glúteos.

Figura 1. Incisiones mediales y laterales de la pierna para una fasciotomía de
cuatro compartimientos. Los compartimientos anterior y lateral se descomprimen
a través de la incisión lateral y los compartimientos superficial y profundo
posterior a través de la incisión medial.

Figura 2. Incisión lateral. Cuando se tienen dudas acerca de la localización de un
compartimiento, una incisión horizontal ayuda a identificar el tabique
intermuscular. El compartimiento anterior descansa en posición anterior a ésta y
el compartimiento lateral lo hace en forma posterior.

Figura 3. Fasciotomía de cuatro compartimientos que puede lograrse mediante
dos incisiones (una medial y una lateral) o una sola incisión lateral.

Figura 4. Fasciotomías del muslo. Los compartimientos anterior y posterior se
descomprimen a través de una incisión lateral y el medial a través de una
incisión medial.
Extremidades superiores
Las incisiones rectas laterales y mediales se utilizan para descomprimir los
compartimientos anteriores y posteriores del brazo, respectivamente (Figura 5).
Una incisión cutánea dorsal y volar se utiliza para el antebrazo (Figura 6). La
incisión dorsal es recta pero la volar debería formar preferentemente una "S-
lenta" o, de forma alternativa, se puede utilizar una incisión volar recta (Figura
7). Para descomprimir los compartimientos de la mano se utilizan cinco incisiones cortas.

Figura 5. Fasciotomías del brazo. El compartimiento anterior se descomprime a
través de una incisión medial y el posterior a través de una incisión lateral.
Obsérvese que los nervios cubital y radial viajan a través de ambos
compartimientos. Así, el incremento en la presión en alguno de los dos producirá
síntomas a lo largo del territorio de distribución de los nervios.
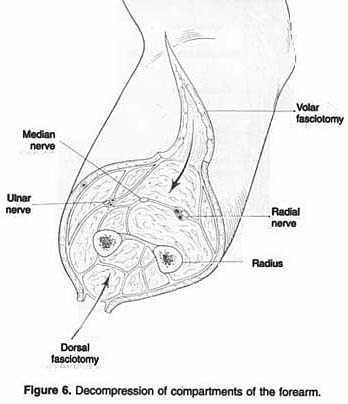
Figura 6. Descompresión de los compartimientos del antebrazo.

Figura 7. Incisiones volar (forma de S o recta) y dorsal (recta) para las fasciotomías
del antebrazo.
Intervenciones farmacológicas
Un número variado de agentes farmacológicos han sido diseńados para
eliminar los radicales libres de oxígeno, para revertir la producción de citoquinas
dańinas y aliviar la lesión por isquemia-reperfusión6 . A pesar de
sus ventajas hipotéticas no existe un beneficio establecido en ensayos humanos.
El manitol se utilizó para prevenir el síndrome compartimental debido a sus
propiedades de reducción de edema y de eliminación de radicales libres.
Prevención de la falla renal
Para prevenir la insuficiencia renal secundaria a rabdomiólisis en pacientes
con síndrome compartimental se emplean la hidratación enérgica junto con
manitol y bicarbonato. La expansión volumétrica temprana es el factor más
significativo para la prevención de la insuficiencia renal aguda.24
En un estudio pequeńo de Homsi y col. no hubo diferencias en los resultados
entre la expansión de volumen con solución salina o con el agregado de manitol
o bicarbonato.25 El manitol contrarresta los tres mecanismos que
se piensa son causantes del deterioro renal en la rabdomiólisis: dilata y
disminuye la viscosidad de los vasos renales incrementando la presión de
perfusión renal; tiene efecto diurético al eliminar acúmulos de los túbulos y tiene
acción antioxidante al eliminarndo el efecto de los radicales libres de la nefrona.
Se piensa que el bicarbonato previene la insuficiencia renal al evitar la
precipitación de los productos de degradación de la mioglobina en los túbulos
mediante la alcalinización de la orina.
A pesar de la ventaja hipotética del manitol/bicarbonato para prevenir la
insuficiencia renal, su acción protectora no está apoyada por la literatura.
Aunque los valores de CPK mayores a 5 000 U incrementan el riesgo de falla
renal, la experiencia con casi 2 000 pacientes con valores anormales de CPK que
fueron internados en la Unidad de Cuidados Intensivos de nuestro centro mostró
que el tratamiento con manitol/bicarbonato no redujo ese riesgo, como tampoco
evitó la necesidad de diálisis ni redujo la mortalidad. Aunque existen algunas
indicaciones de que podría ser beneficioso en pacientes con valores de CPK
mayores a 30 000 U.26
Complicaciones del síndrome compartimental
Se podría pensar en complicaciones locales o
sistémicas.1,2,9,16,23 Las locales incluyen infección y necrosis
muscular. Para prevenir las infecciones, las fasciotomías y los siguientes cambios
de vendajes deberían realizarse de forma estéril. El músculo necrótico se
convierte gradualmente en tejido rígido, lo que genera contracturas de Volkmann
si se dejan sin terapia física.
Las complicaciones sistémicas pueden estar dirigidas hacia cualquier órgano. La
insuficiencia renal, el síndrome de distrés respiratorio del adulto, la insuficiencia
cardíaca y la coagulación intravascular diseminada son las complicaciones
sistémicas más frecuentemente halladas. El riesgo para su desarrollo es
proporcional a la cantidad de sustancias tóxicas liberadas a partir del músculo
isquémico, lo que es proporcional a la masa muscular y a la duración de la
isquemia.
Los autores no manifiestan conflictos.
BIBLIOGRAFÍA
-
Matsen FA III. Compartmental syndromes. New York, Grune and Stratton,
1980;pp 1-162.
-
Mubarak SJ, Hargens AR. Compartment syndromes and Volkmann's
contracture. Philadelphia, WB Saunders, 1981, pp 1-232.
-
Harris K, Walker PM, Mickle DAG, et al. Metabolic response of skeletal muscle
to ischemia. Am J Physiol 1986;250:213-20.
-
Moore RE, III, Friedman RJ. Current concepts in pathophysiology and diagnosis
of compartment syndrome. J Emerg Med 1989;7:657-62.
-
Davies MG, Hagen PO. The vascular endothelium. A new horizon. Ann Surg
1993;218:593-609.
-
Allen DM, Chen L, Seaber AV, et al. Pathophysiology and related studies of the
no reflow phenomenon in skeletal muscle. Clin Orthop 1995;314:122-33.
-
Ames A, Wright RL, Kowada M, et al. Cerebral ishemia – the no reflow
phenomenon. Am J Pathol 1968;52:437-53.
-
Granger DN. Role of xanthine oxidase and granulocytes in ischemia-reperfusion
injury. Am J Physiol 1988;1363-6135:H1269-H1275.
-
Grishame MB, Jefferson MM, Thomas EL. Role of monochloramine in the
oxidation of erythrocyte hemoglobin by stimulated neutrhophils. J Biol Chem
1984;259:6757-65.
-
Petrone WF, English DK, Wong K, et al. Free radicals and inflammation:
Superoxide-dependent activation of a neutrophil chemotactic factor in plasma. Proc
Natl Acad Sci 1979;77:1159-63.
-
Hofmeister EP, Shin AY. The role of prophylactic fasciotomy and medical
treatment in limb ishemia and revascularization. Hand Clin N Am 1998;14:457-65.
-
Yassin MM, Harkin DW, Barros D'Sa AA, et al. Lower limb ischemia-reperfusion
injury triggers a systemic inflammatory response and multiple organ dysfunction.
World J Surg. 2002;1:115-21.
-
Cywes S, Louw JG. Phlegmasia cerulea dolens: successful treatment by
relieving fasciotomy. Surgery 1962;51:169-72.
-
Dennis C. Disaster following femoral vein ligation for thrombophlebitis: relief by
fasciotomy. Surgery 1945;17:264-9.
-
Gulli B, Templeman D. Compartment syndrome of the lower extremity. Orthop
Clin N Am 1994;25:677-84.
-
Hill SL, Bianchi J. The gluteal compartment syndrome. Am Surg 1997;9:823-6.
-
Doyle J. Anatomy of the upper extremity muscle compartments. Hand Clin N
Am 1998;14:343-64.
-
Velmahos GC, Vassiliu P. Extremity compartment syndrome. In: Trauma
Management. D Demetriades, JA Asensio (eds). Landes Bioscience 2000, pp 405-
12.
-
Hargens AR, Akeson WH, Mubarak SJ, et al. Interstitial fluid pressure in muscle
and compartment syndrome in man. Microvasc Res 1977;14:1-10.
-
Matsen FA, III, Mayo KA, Sheridan GW, Krugmire RB, Jr. Monitoring of
intramuscular pressure. Surgery 1976;79:702-9.
-
Rorabek CH, Clarke KM. The pathophysiology of the anterior tibial compartment
syndrome: an experimental investigation. J Trauma 1978;18:299-304.
-
Sheridan GW, Matsen FA, III, Krugmire RB, Jr. Further investigation on the
pathophysiology of the compartmental syndrome. Clin Orthop Relat Res
1977;123:266-70.
-
Hargens AR, Mubarak SJ. Current concepts in the pathophysiology, evaluation,
and diagnosis of compartment syndrome. Hand Clin N Am 1998;14:371-83.
-
White TO, Howell GE, Will EM, et al. Elevated intramuscular compartment
pressures do not influence outcome after tibial fracture. J Trauma. 2003;55:1133-
8.
-
Robbs JV, Baker LW. Late revascularization of the lower limb following acute
arterial occlusion. Br J Surg 1979;78:490-3.
-
Oda J, Tanaka H, Yoshioka T, et al. Analysis of 372 patients with crush
syndrome caused by the Hanshin-Awaji earthquake. J Trauma 1997;42:470-6.
-
Vanholder R, Sever MS, Erek E, et al. Rhabdomyolysis. J Am Soc Nephrol.
2000;11:1553-61
-
Lappalainen H, Tiula E, Uotila L, et al. Elimination kinetics of myoglobin and
creatine kinase in rhabdomyolysis: implications for follow-up. Crit Care Med.
2002;30:2212-5.
-
Ward MM. Factors predictive of acute renal failure in rhabdomyolysis. Arch
Inten Med 1988;148:15537.
-
Knottenbelt JD. Traumatic rhabdomyolysis from severe beating-Experience of
volume diuresis in 200 patients. J Trauma 1994;37:214-9.
-
Nimmo GR, Lambie AT, Cumming AD. Rhabdomyolysis and acute renal failure.
Intens Care Med 1989;15:486-7.
-
Mars M, Hadley GP. Failure of pulse oximetry in the assessment of raised limb
intracompartmental pressure. Injury 1994;25:379-81.
-
Giannoti G, Cohn SM, Brown M, et al. Utility of near-infrared spectroscopy in
the diagnosis of lower extremity compartment syndrome. J Trauma 2000;48:396-
401.
-
Garr JL, Gentilello LM, Cole PA, et al. Monitoring for compartmental syndrome
using near-I spectroscopy: A noninvasive, continuous, transcutaneous monitoring
technique. J Trauma 1999;46:613-8.
-
Gentilello LM, Sanzone A, Wang L, et al. Near-infrared spectroscopy versus
compartment pressure for the diagnosis of lower extremity compartmental
syndrome using electromyography-determined measurements of neuromuscular
function. J Trauma. 2001;51:1-8.
-
Shah PM, Wapnir I, Babu S, et al . Compartment syndrome in combined arterial
and venous injury of the lower extremity. Am J Surg 1989;158:136-41.
-
McGee DL, Dalsey WC. The mangled extremity. Compartment syndrome and
amputations. Emerg Clin N Am 1992;10:783-800.
-
Perry MO. Compartment syndromes and reperfusion injury. Surg Clin N Am
1988;68:853-68.
-
Velmahos GC, Theodorou D, Demetriades D, et al. Complications and
nonclosure rates of fasciotomy for trauma and related risk factors. World J Surg
1997;21:247-53.
-
Matsen FA, Krugmire RB. Compartment syndromes. Surg Gynecol Obstet
1978;147:943-9.
-
Hallock GG. An endoscopic technique for decompressive fasciotomy. Ann Plast
Surg 1999;43:668-7
-
Herbert KJ, Hickey MJ, Lepore DA, et al. Effects of the endothelin receptor
antagonist Bosentan on ischaemia/reperfusion injury in rat skeletal muscle. Eur J
Pharmacol. 2001 Jul 13;424:59-67.
-
Ron D, Taitelman U, Michaelson M, et al. Prevention of acute renal failure in
traumatic rhabdomyolysis. Arch Intern Med. 1984 Feb;144:277-80.
-
Homsi E, Barreiro MF, Orlando JM, et al. Prophylaxis of acute renal failure in
patients with rhabdomyolysis. Ren Fail. 1997 Mar;19:283-8.
-
Brown CV, Rhee P, Chan L, et al. Preventing renal failure in patients with
rhabdomyolysis: Do bicarbonate and mannitol make a difference? J Trauma (in
press).
-
Lagerstorm CF, Reed LR, Rowlands BJ, et al. Early fasciotomy for acutre
clinically evident post-traumatic compartment syndrome. Am J Surg 1989;158:36-
40.
-
Filed CK, Senkowsky J, Hollier LH, et al. Fasciotomy for vascular trauma: is it
too much, too often? Am Surg 1994;60:409-13.
-
Fitzgerald AM, Gaston P, Wilson Y, et al. Long-term sequelae of fasciotomy
wounds. Br J Plast Surg 2000;53:690-3.
-
Harris I. Gradual closure of fasciotomy wounds using a vessel loop shoelace.
Injury 1993;24:565-7.
-
Almekinders LC. Gradual closure of fasciotomy wounds. Orthop Rev
1991;20:82-4.
-
Hirvensalo E, Tuominen H, Lapinsuo M, et al. Compartment syndrome of the
lower limb caused by a tourniquet: A report of two cases. J Orthop Trauma
1992;4:469-72.
|



