Introducción
La leucemia linfoblástica aguda (LLA) es el cáncer mas común en la infancia. Con
una frecuencia de 3.3 casos por cada 100 000 nińos, constituye el 30% de todos los
cánceres en esa edad.1 La variedad más frecuente es la que se
origina en los linfocitos B, que comprende casi el 90% de los casos.1
Con tratamiento óptimo la tasa de curación esta aproximándose al 80% en los
países con mayor desarrollo en sus sistemas de salud.2 La L-
asparaginasa es parte fundamental del tratamiento, se la incluye en diversas dosis
en todos los esquemas terapéuticos.3,4 Su uso intensivo ha
resultado en mejores tasas de respuesta,5,6 aunque puede
acompańarse de formación de anticuerpos anti-asparaginasa7 y de
reacciones de hipersensibilidad, que pueden o no implicar el desarrollo de
resistencia al efecto de la enzima,8 pero que obligan a cambiar a
una asparaginasa de origen diferente a la de E. coli, como la obtenida de
Erwinia carotobora.
La administración de L-asparaginasa puede acompańarse, además, de eventos
tromboembólicos graves, los cuales se presentan con una incidencia del 2.4% al
36.7%.9,10 Diversos estudios documentaron una disminución del
fibrinógeno y de las proteínas anticoagulantes C, S y antitrombina III (ATIII) en
nińos que reciben tratamiento para la LLA.11,12 Recientemente se
identificaron mutaciones en estas proteínas, así como en la protrombina y el factor
V.9 Sin embargo éstas tienen la prevalencia esperada en la
población, por lo que por sí mismas no explican los eventos tromboembólicos, de lo
que se deduce la existencia de factores procoagulantes adicionales relacionados al
tratamiento, factores ambientales o manbos.9,12 En esta
investigación postulamos la hipótesis de que la L-asparaginasa tiene un efecto
agonista plaquetario, que podría contribuir al estado hipercoagulable y al riesgo de
desencadenar eventos trombóticos en nińos con LLA.
Pacientes y métodos
En el estudio se siguieron los principios establecidos en la Declaración de
Helsinki y se obtuvo el consentimiento informado de ambos padres. El protocolo fue
aprobado por el Comité de Etica de la institución. Se estudiaron veinte pacientes
menores de 12 ańos con el diagnóstico de LLA de la infancia de riesgo estándar,
sometidos a un protocolo de inducción de la remisión que incluyó vincristina, 1.4
mg/m2/i.v., prednisona, 60 mg/m2/día/v.o., y L-
asparaginasa 6 000 U/m2 / 2 veces por
semana. El grupo control incluyó 15 individuos sanos, no fumadores, sin
ingestión de anti-inflamatorios no esteroides y con tiempo de sangrado y cuenta de
plaquetas normales.
En los pacientes los estudios de agregometría plaquetaria se realizaron durante la
fase de inducción de la remisión, cuando se habían recibido al menos cuatro dosis
de L-asparaginasa y la cuenta de plaquetas era > 100x109/l,
utilizando un método publicado previamente.13 Se empleó un
agregómetro óptico automatizado (Chronolog modelo 530VS, Haverton, PA,
EE.UU.) que lleva a cabo la integración del proceso por medio de una interfase y
software específicos (Aggrolink, Haverton, PA, EE.UU.).
Se obtuvieron 20 ml de sangre venosa, descartando los primeros 5 ml, sin
aplicación de torniquete o utilizando éste con una compresión ligera, en tubos de
plástico conteniendo citrato de sodio al 3.8%, en una relación 1:9 v/v. La muestra
se obtuvo dentro de las 24 horas posteriores a la administración de la cuarta dosis
de L-asparaginasa, cuya vida media es de 26 horas.14 Las plaquetas
se obtuvieron preparando un plasma rico en plaquetas a 135 g por 15 minutos a
temperatura ambiente. Durante una segunda centrifugación a 1 500 g por 15
minutos se obtuvo el plasma pobre en plaquetas para emplearlo en la dilución y
ajuste de la muestra a una cuenta de 250 000 plaquetas x 109/l, y
para establecer la lectura basal de agregación en 0%. Se estudiaron las respuestas
a 37°C a 1 000 revoluciones por minuto (rpm) a los siguientes
agonistas:13 ristocetina, 1.5 mg/ml, adenosin difosfato (ADP) 10
μM, epinefrina 10 μM y colágeno 2.5 μg/ml. Diluciones salinas de L-
asparaginasa (Leunase, Kyowa Hakko Kogyo Co. Ltd, Tokio, Japón) en
concentraciones de 2 500, 5 000, y 10 000 unidades en solución fisiológica al 0.9%,
con un pH de 7.3, fueron empleadas como agonistas en ensayos independientes. Se
agregó cada agonista en un volumen de 50 μl a una cubeta conteniendo 450
μl de la muestra y se procedió al ensayo; cada ensayo incluyó un control de
voluntarios normales. El resultado, expresado como porcentaje de agregación, se
analizó en la gráfica correspondiente. Se procedió a comparar las diferentes curvas
generadas.
Resultados
Las características clínicas e iniciales de laboratorio de los nińos con LLA,
expresados como la mediana y el rango, incluyeron las siguientes: edad: 6.0 ańos
(2-13); hemoglobina: 62 g/l (48-89); leucocitos totales: 23 200 por microlitro
(948-105 000); plaquetas: 31 300 por microlitro (1 170-259 000); albúmina: 3.5
g/l (2.4-4.8); proteínas totales: 6.5 g/l (5.9-7.5); inmunoglobulina G sérica: 945
mg/dl (400-1 209). La agregometría demostró un claro efecto proagregante de la
L-asparaginasa, tanto en plaquetas de pacientes con LLA en fase de inducción
(figura 1), como en las de voluntarios normales (no mostrada). El porcentaje de
agregación con las diluciones de la L-asparaginasa varió de 28% con una
concentración de 2 500 UI, 52% cuando se empleó la concentración de 5 000 UI
(figura 1), hasta un 100%, en el caso de la dilución de 10 000 UI, presentándose
como una potente onda de agregación primaria en menos de un minuto del ensayo.
El porcentaje de desagregación fue menor al 5% a los 7 minutos de registro. Se
apreció una correlación entre la concentración de la L-asparaginasa y el porcentaje
de agregación plaquetaria en ambos grupos. No se detectaron diferencias en la
agregometría entre las plaquetas de individuos sanos y de pacientes con LLA, ni con
agonistas usuales ni con las diferentes concentraciones de L-asparaginasa; en la
figura 2 se muestra el resultado normal de la agregometría efectuada en el mismo
paciente utilizando colágeno, 2.5 microgramos por ml.

Figura 1. Agregación plaquetaria (52%) determinada en plasma rico en plaquetas de un paciente con
leucemia aguda linfoblástica de la infancia, después de estimulación con una dilución de L-asparaginasa
de 5 000 UI/ml. Es de notarse la velocidad acelerada de la reacción durante los primeros segundos,
progresando después lentamente hasta alcanzar una fase de meseta ligeramente superior al 50% de la
agregación.
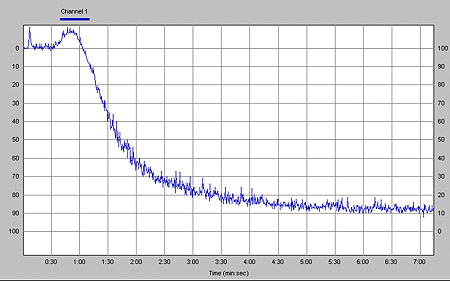
Figura 2. Agregación plaquetaria (90%) determinada en plasma rico en plaquetas por agregometría
óptica en el mismo paciente de la figura 1, después de estimulación con un reactivo estándar de
colágeno, 2.5 μg/ml.
Uno de los pacientes, una nińa de 4 ańos, desarrolló trombosis en el hemisferio
cerebral derecho, documentada por tomografía axial computarizada, después de la
administración de la tercera dosis de L-asparaginasa, cuando su cuenta plaquetaria
era de solamente 20x109/l. Su patrón de respuesta en la
agregometría resultó similar al de los otros pacientes, con respuesta de agregación
in vitro al estímulo de la L-asparaginasa.
Discusión
La disminución de las proteínas anticoagulantes C, S y ATIII ha sido
reconocida como una de las causas principales del estado hipercoagulable en nińos
con LLA.9 Dicho estado puede conducir a eventos trombóticos graves
y se cree que es el resultado de la quimioterapia administrada,11 y
de otros factores todavía no identificados.9 Como ejemplo, en un
estudio reciente se encontró una asociación entre el desarrollo de eventos
tromboembólicos y la aparición de anticuerpos antifosfolípidos, más que con la
presencia de los factores protrombóticos hereditarios previamente
citados.10 La naturaleza inherentemente compleja de este estado de
desequilibrio hemostático en la LLA, así como los múltiples factores que pueden
influir en el desencadenamiento de eventos trombóticos, ha sido recientemente
revisada, contemplándose la posible participación de, entre otros, la liberación de
micropartículas celulares conteniendo factor tisular activado, así como la inducción
de formación de trombos secundaria a infección bacteriana.15
La L-asparaginasa en dosis elevadas forma parte integral del tratamiento exitoso de
la LLA de la infancia y se la ha relacionado con el estado hipercoagulable durante la
fase de inducción de la remisión16 y con el desarrollo de anticuerpos
específicos y reacciones de hipersensibilidad17 que obligan a utilizar
una asparaginasa de origen diferente del de E. coli,8 un
protocolo de desensibilización o ambas medidas.18 La L-
asparaginasa ha sido estudiada con relación al desarrollo del estado hipercoagulable
de la LLA en asociación con la disminución de las proteínas C, S y ATIII. Sin
embargo, la L-asparaginasa no ha sido, hasta donde sabemos, estudiada o
involucrada en la activación de la fase plaquetaria de la coagulación ni empleada
como agonista en estudios de los efectos proagregantes de otros agentes.
En este estudio se demostró claramente que la L-asparaginasa puede actuar como
un potente agonista plaquetario in vitro, en forma proporcional a su
concentración, lo mismo en plaquetas de nińos con LLA que en las de individuos
sanos, libres de patología plaquetaria. Aunque este efecto proagregante no ha sido
documentado in vivo, su administración podría ser uno de los factores
protrombóticos no identificados relacionados con el tratamiento, y cuya existencia
se sugirió en estudios previos.9 En conjunto con la disminución de
las proteínas anticoagulantes naturales, un efecto proagregante de la enzima podría
contribuir al estado hipercoagulable observado en la fase inicial del tratamiento de
la LLA.11
Recientemente se informó que el alcaloide de la vinca, vincristina, utilizado junto
con la L-asparaginasa en la fase de inducción de la remisión de la LLA, es capaz de
inducir apoptosis in vivo de células mononucleares malignas, con un índice
apoptótico de hasta 40%.19 La apoptosis participa en un numeroso
grupo de fenómenos biológicos, entre ellos, de manera notable, la trombogénesis,
probablemente a través de la activación del factor tisular,20 se sabe
que la interacción de las plaquetas con las células mononucleares en otras
enfermedades, como el lupus eritematoso sistémico, acelera la producción del
factor tisular por los monocitos activados,21 algo similar podría
suceder por medio de la interacción entre plaquetas y linfoblastos apoptóticos. La
formación de estos complejos heterotípicos circulantes, que podrían contribuir al
estado hipercoagulable, ha sido documentada entre plaquetas y
neutrófilos,22 y entre plaquetas y células
mononucleares,23 por lo que cabe la posibilidad de su formación
entre plaquetas y linfoblastos leucémicos.
Es posible que un estado de agregación plaquetaria aumentada debido a un efecto
proagregante asociado con la administración de L-asparaginasa, como se
documentó in vitro en este estudio, en combinación con la disminución en
la síntesis de las proteínas anticoagulantes naturales C, S y ATIII, aunada a los
fenómenos apoptóticos procoagulantes secundarios a la utilización de vincristina,
ayude a explicar de manera más completa el desencadenamiento de fenómenos
trombóticos en la fase de inducción de la remisión de la LLA de la infancia. Es
también evidente, a la luz de éstos y otros hallazgos, que diferentes protocolos
para el tratamiento de la LLA pueden conducir a diferentes tasas de
tromboembolismo,9,10 probablemente debido a diferencias en las
dosis, esquemas y rutas de administración de los agentes quimioterapéuticos. A
este respecto, las combinaciones de los diferentes agentes utilizados,
particularmente cuado se emplea prednisona en sustitución de la dexametasona
durante la fase de inducción de la remisión, puede conducir a diferencias
dramáticas en la incidencia de eventos trombóticos.24 Una
agregometría plaquetaria ha sido previamente sugerida en pacientes con trastornos
mieloproliferativos y eventos trombóticos25 y podría estar indicada,
utilizando diluciones de la L-asparaginasa como agonista adicional, para
identificar el subgrupo de nińos en riesgo de sufrir eventos trombóticos, con el
fin de evaluar la administración de anticoagulantes de manera profiláctica, como se
ha propuesto para el uso de la heparina de bajo peso molecular en aquellos
pacientes en los que se identifica una disminución significativa de proteínas
anticoagulantes C, S y ATIII.9 Adicionalmente, debe estudiarse la
posibilidad de que la enzima contribuya de manera directa a la deficiencia de estas
proteínas, ya que se ha documentado la disminución de la actividad de diversas
proteínas involucradas en el control de la coagulación después de breves intervalos
de incubación del plasma con diversas concentraciones de L-
asparaginasa.26
Los autores no manifiestan conflictos
BIBLIOGRAFÍA
-
Pui CH. Acute lymphoblastic leukemia, in Williams Hematology,
6th ed, Beutler E, Lichtman M, Coller B, and Selighson U (eds), p
1141-1161. McGraw Hill, New York, 2001.
-
Greaves M. Science, Medicine and the Future. Childhood leukemia. Br Med J
2002;324:283-7.
-
Felix CA, Lange BJ, Chessells JM. Pediatric acute lymphoblastic leukemia.
Hematology 2000;**:285-302.
-
Richards S, Burrett J, Hann I, Chessells J, Hill F, Bailey C. Improved survival
with early intensification: combined results from the Medical Research Council
childhood ALL randomized trials, UKALL X and UKALL XI. Medical Research Working
Party on Childhood Leukemia. Leukemia 1998;12:1031-1036.
-
Nachman JB, Sather HN, Sensel MG, Triggh ME, Cherlow JM, Lukens JN.
Augmented post-induction therapy for children with high-risk acute lymphoblastic
leukemia and a slow response to initial therapy. N Eng J Med 1998;338:1663-1671.
-
Amylon MD, Shuster J, Berard C, Link MP, Wharam M. Intensive high-dose
asparaginase consolidation improves survival for pediatric patients with T cell acute
lymphoblastic leukemia and advanced stage lymphoblastic lymphoma: a Pediatric
Oncology Group study. Leukemia 1999;13:335-342.
-
Woo MH, Hak LJ, Storm MC, Sandlund JT, Rivera GK, et al. Anti-asparaginase
antibodies following E. coli asparaginase therapy in children with acute
lymphoblastic leukemia. Leukemia 1998;12:1257-1533.
-
Larson RA, Fretzin MH, Dodge RK, Schiffer CA. Hypersensitivity reactions to L-
asparaginase do not impact on the remission duration of adults with acute
lymphoblastic leukemia . Leukemia 1998;12:660-665.
-
Nowak-Gotti U, Wermes C, Junker R, Koch HG, Schobess R, Fleischhack G.
Prospective evaluation of the thrombotic risk in children with acute lymphoblastic
leukemia carrying the MTHFR TT 667 genotype, the prothrombin G20210A variant,
and further prothrombotic risk factors. Blood 1999;93:1595-1599.
-
Mitchell L, Andrew M, Hanna K, Abshire T, Chait P, Halton J, et al. A prospective
cohort study determining the prevalence of thrombotic events in children with acute
lymphoblastic leukemia and a central venous line who are treated with L-
asparaginase: results of the Prophylactic Antithrombin Replacement in Kids with
ALL Treated with Asparaginase Group (PARKAA). Cancer 2003;97:508-16.
-
Mitchell LG, Sutor AH, Andrew M. Hemostasis in childhood acute lymphoblasitic
leukemia : coagulopaty induced by disease or treatment. Semin Thromb Hemost
1995;21:390-401.
-
Corso A, Castagnola C, Bernasconi C. Thrombotic events are not exclusive to
the remission induction period in patients with acute lymphoblastic leukemia: a
report of two cases of cerebral sinus trombosis. Ann Hematol 1997;75:117-119.
-
McCabe WM, Jennings LK. Laboratory evaluation of platelet function, in:
Platelet Protocols, Research and Clinical Laboratory Procedures, McCabe WM and
Jennings LK, (eds) p. 40-50. Academic Press, San Diego, CA, 1999.
-
Avramis VI, Sencer S, Periclou AP, sather H, Bostrom BC, Cohen LJ, Ettinger
AG, Ettinger LJ, franklin J, gaynon OS, Hilden JM, Lange B, Majlessipour F, Mathew
P, Needle M, Neglia J, Reaman G, Holsenberg JS. A randomized comparison of
native Escherichia coli asparaginase and polyethylene glycol conjugated
asparaginase for treatment of children with newly diagnosed standard-risk acute
lymphoblastic leukemia: a Children´s Cancer Group study. Blood 2002;99:1986-
1994.
-
Jaime-Pérez JC, Gómez-Almaguer D. The complex nature of the prothrombotic
state in acute lymphoblastic leukemia of childhood. Haematologica
2003;88:(07)ELT25
-
Castaman G, Rodeghiero F, Dini E. Thrombotic complications during L-
asparaginase treatment for acute lymphoblastic leukemia. Haematologica
1990;75:567-569.
-
Woo MH, Hak LI, Storm MC, Sandlund JT, Ribeiro RC, Rivera GK, Rubnitz JE,
Harrison PL, wang B, Evans WE, Pui CH, Relling MV. Hypersensitivity or
development of antibodies to asparaginase does not impact treatment outcome of
acute lymphoblastic leukemia. J Clin Oncol 2000;18:1525-1532.
-
Bonno M, Kawasaki H, Hori H, Umemoto M, Sakurai M,. Rapid desensitization
for L-asparaginase sensitivity. J Aller Clin Immunol 1998;101:497-502.
-
Groninger E, de Graaff SSN, Meeuwesen-de-Boer GJ, Sluiter WJ, Poppema S.
Vincristine induced apoptosis in vivo in peripheral blood mononuclear cells of
children with acute lymphoblasatic leukemia. Br J Haematol 2000;111:875-878.
-
Wang J, Weiss I, Svoboda K, Kwaan C. Thrombogenic role of cells undergoing
apoptosis. Br J Haematol 2001;115:382-391.
-
Silverstein RL, Nachman RL. Thrombospondin binds to monocytes-
macrophages and mediates platelet-monocyte adhesion. J Clin Invest 1987;79:867-
874.
-
Peters MJ, Dixon G, Kotowicz KT, Hatch DJ, Heyderman RS, Klein NJ.
Circulating platelet-neutrophil complexes represent a subpopulation of activated
neutrophils primed for adhesion, phagocytosis and intracellular killing. Br J
Haematol 1999:106:391-399.
-
Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, Wagner DD, Furie
B. PADGEM protein: a receptor that mediates the interaction of activated platelets
with neutrophils and monocytes. Cell 1989; 59:305-312.
-
Nowak-Göttl U, Alkhe E, Fleischhack G, Schwabe D, Schobess R, Schumann C,
Wermes C, Junker R. Thromboembolic events in children with acute lymphoblastic
leukemia (BFM protocols): prednisone versus dexamethasone administration. Blood
2003;101:25-29-33.
-
Manoharan A, Gemmell R, Brighton T, Dunkley S, Lopez A, Kkyle P. Thrombosis
and bleeding in myeloproliferative disorders: identification of at-risk patients with
whole blood platelet aggregation studies. Br J Haematol 1999;105:618-625.
-
Nowak-Gottl U, Boos J, Wolff JE, Lill H, Veltmann H, Werber G, Ahlke E,
Jurgens H. Asparaginase decreases clotting factors in vitro: a possible pitfall? Int J
Clin Lab Res. 1995;25(3):146-8.
|