Introduçăo
O aumento do fígado é um achado clínico comum na infância, podendo ser decorrente de doença hepática intrínseca ou de alteraçőes sistęmicas.1 A determinaçăo do tamanho do fígado é, portanto, procedimento de rotina no exame clínico de crianças.
O método clínico de biometria hepática, contudo, apresenta limitaçőes, decorrentes de variaçőes na forma, eixo e posiçăo do fígado,2,3 de suas relaçőes com estruturas vizinhas2,4 e da técnica empregada,5,6 sendo operador dependente.7
A hepatometria “in vivo” pode, ainda, ser realizada por métodos de diagnóstico por imagem, tais como: radiografia,9,10-14 cintilografia,3,15- 20 ultra-sonografia15,20-38 e tomografia computadorizada,39 que, por sua vez, procuram oferecer subsídios mais concretos, com melhor reprodutibilidade, com o objetivo, tanto de complementar a avaliaçăo clínica, como de minimizar o fator operador-dependęncia deste procedimento.
Dentre os métodos de imagem, a ultra-sonografia apresenta vantagens, sobretudo na avaliaçăo do paciente pediátrico, por tratar-se de método totalmente năo invasivo, năo utilizar radiaçăo ionizante e năo exigir sedaçăo, além de permitir o estudo minucioso do paręnquima e estruturas hepáticas. Assim, a ultra-sonografia é, de modo geral, o primeiro exame de imagem solicitado para esclarecimento diagnóstico, quando há suspeita clínica de hepatomegalia e o exame de escolha no monitoramento de alteraçőes do tamanho do fígado, que possam ocorrer na evoluçăo natural de doenças ou em conseqüęncia a terapia.
O método ultra-sonográfico, no entanto, tem como limitaçőes o fator operador- dependęncia e as barreiras ŕ progressăo do feixe sonoro, representadas pelo ar e estruturas ósseas. As barreiras ao som assumem especial relevância no que se refere ao estudo do fígado, devido a sua localizaçăo, uma vez que, o acesso ao órgăo pode ser prejudicado pela interposiçăo dos arcos costais e porçăo variável de paręnquima pulmonar que se insinua entre o fígado e a parede abdominal.
A forma do fígado constitui outro fator de dificuldade para a padronizaçăo de medidas, pois, sendo bizarra, grosseiramente semelhante a uma cunha e com ampla superfície superior, arredondada, exige definiçăo de pontos de referęncia precisos quando se pretende padronizar uma técnica com boa reprodutibilidade.
No que se refere ŕ hepatometria ultra-sonográfica em crianças, há multiplicidade de métodos propostos,21,24-26,28,33,35 o que, a nosso ver, revela a falta de um, que atenda ŕs necessidades do examinador, isto é, um método preciso, reprodutível e de fácil execuçăo.
Com o intuito de determinarmos um método que atendesse a essas prerrogativas, realizamos estudo de mensuraçăo do fígado de crianças, entre 0 e 6 anos de idade,40 em que avaliamos diversos parâmetros de hepatometria, quanto ŕ reprodutibilidade inter-observador e quanto ŕs dificuldades técnicas para sua obtençăo. Nesse trabalho foi realizada, ainda, a análise de correlaçăo dos diversos parâmetros entre si.
Método
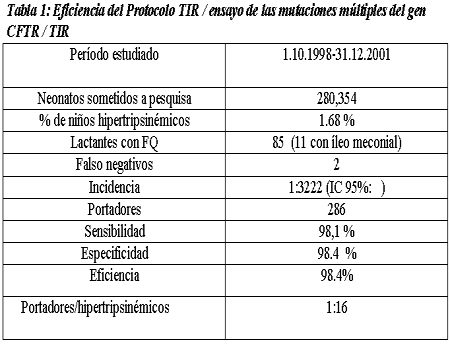
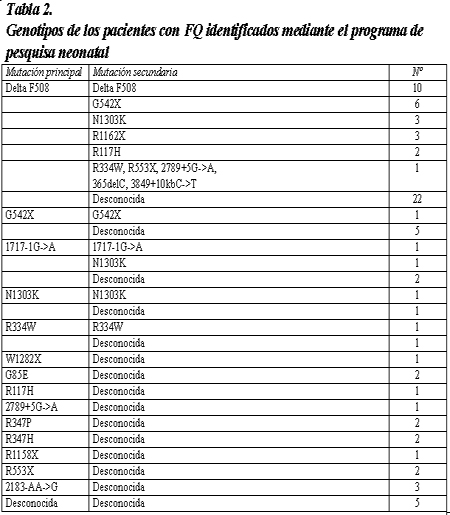
Propomos método de biometria hepática baseado em dois planos seccionais longitudinais, estabelecidos pela inter-relaçăo de reparos anatômicos intra- abdominais, intra e extra-hepáticos. Para a avaliaçăo do tamanho do fígado săo utilizados dois parâmetros: a) o diâmetro crânio-caudal do lobo hepático esquerdo, medido na linha médio esternal (LME), tendo como reparo anatômico intra-hepático a veia hepática esquerda e b) o diâmetro crânio-caudal posterior do lobo hepático direito, na linha hemi-clavicular (LHC), tendo como reparo anatômico intra-hepático o ramo portal direito (figuras 1, 2 e 3).
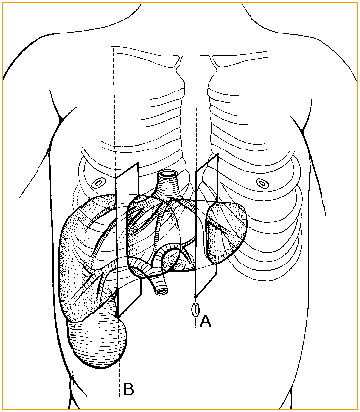
Figura1. Representaçăo esquemática dos planos de corte para a hepatometria em crianças, estabelecidos pelas linhas de orientaçăo externas: A (linha médio-esternal, para a medida do lobo hepático esquerdo) e B (linha hemi-clavicular, para a medida do lobo hepático direito).
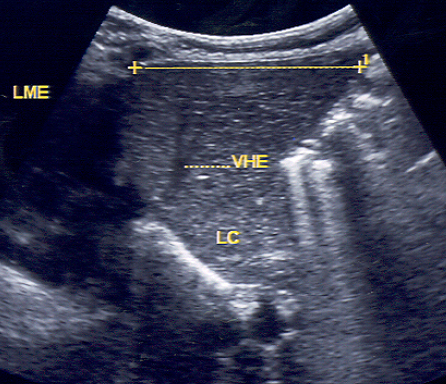
Figura 2. Medida do diâmetro crânio-caudal do lobo hepático esquerdo na linha médio-esternal (LME), tendo como reparo anatômico intra-hepático a veia hepática esquerda (VHE). Ressalta-se que a veia cava inferior năo deve ser incluída na imagem. LC: lobo caudado.
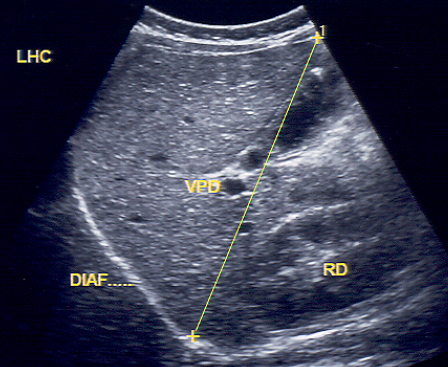
Figura 3. Medida do diâmetro crânio-caudal posterior do lobo hepático direito, medido na linha hemi- clavicular (LHC), tendo com reparo anatômico intra-hepático a veia porta direita (VPD), em corte transversal. Reparos anatômicos intra-abdominais extra-hepáticos: rim direito (RD) e ampla visualizaçăo do diafragma.
Comentários
O método proposto foi composto pelos parâmetros que apresentaram menores dificuldades técnicas e altos índices de correlaçăo positiva com os demais parâmetros estudados, de forma a poderem representá-los. Ressalta-se que o diâmetro antero-posterior do lobo hepático esquerdo năo apresentou correlaçăo com qualquer dos parâmetros analisados.
O método se mostrou reprodutível por um mesmo observador e a introduçăo de reparos anatômicos intra-hepáticos resultou proveitosa, permitindo definiçăo mais precisa dos planos de corte para aferiçăo das medidas, minimizando o fator operador-dependęncia.
Estudo da variabilidade inter-observador
No período entre Agosto e Outubro de 2002 foi realizado estudo com o objetivo de testar a reprodutibilidade inter-observador do novo parâmetro introduzido (diâmetro crânio-caudal posterior na linha hemi-clavicular - CCPLHC) e do parâmetro classicamente utilizado para a medida do lobo hepático direito (diâmetro crânio-caudal anterior na linha hemi-clavicular - CCALHC).
Populaçăo de estudo e metodo
Tręs examinadores independentes realizaram a biometria hepática em 31 crianças, com idades entre 0 e 6 anos, aplicando método ultra-sonográfico padronizado. Todas as crianças haviam sido encaminhadas para exame de ultra- sonografia abdominal no Setor de Diagnóstico por Imagem do ICR devido a suspeitas diagnósticas diversas. Foram excluídas da amostra crianças com doenças crônicas hepáticas ou das vias biliares.
Os exames de ultra-sonografia foram realizados em Modo-B, com equipamento ATL modelo APOGEE 800 PLUS e Aloka modelo SS-2000, utilizando-se transdutor convexo de 5.0 MHz.
A biometria hepática foi realizada separadamente por cada um dos examinadores (observador 1, 2 e 3), sem uma ordem pré-definida. Foram medidos os diâmetros CCPLHC e CCALHC.
A variabilidade inter-observadores foi testada utilizando-se o coeficiente de correlaçăo de Pearson e o modelo de regressăo linear.
Resultados
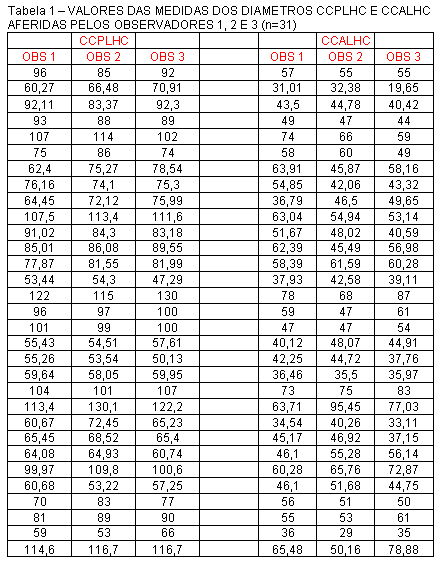
Os valores das medidas dos diâmetros CCPLHC e CCALHC obtidos pelos observadores 1, 2 e 3 săo apresentados na tabela 1.
Os resultados do estudo de correlaçăo mostraram que houve correlaçăo positiva e significante entre os valores das medidas obtidas pelos tręs observadores para ambos os parâmetros analisados, ou seja, para um mesmo parâmetro, quanto maior o valor obtido por um observador, maior o valor obtido pelo outro.
Aplicando-se o modelo de regressăo observou-se que os observadores 1, 2 e 3 reproduziram os valores de CCPLHC, enquanto que năo houve reproduçăo dos valores de CCALHC por qualquer dos observadores.
Nos gráficos 1 a 6 săo apresentados os coeficientes de correlaçăo de Pearson (r), o nível de significância (p) e os modelos de regressăo linear para os valores de CCPLHC (gráficos 1, 2 e 3) e CCALHC (gráficos 4, 5 e 6).
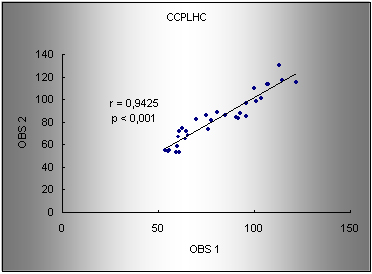
Gráfico 1. Regressăo para os valores de CCPLHC entre os observadores 1 e 2. r: coeficiente de correlaçăo de Pearson. Modelo de regressăo: obs1 = 5.65 + 0.91 (obs2).
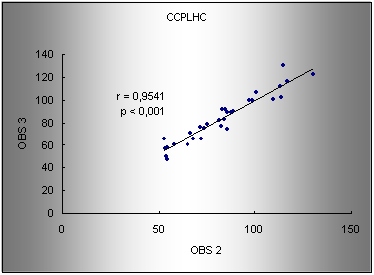
Gráfico 2. Regressăo para os valores de CCPLHC entre os observadores 2 e 3. r: coeficiente de correlaçăo de Pearson. Modelo de regressăo: obs2 = 3.26 + 0.96 (obs3).
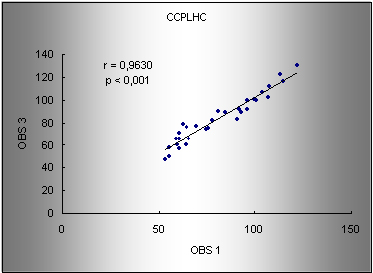
Gráfico 3. Regressăo para os valores de CCPLHC entre os observadores 1 e 3. r: coeficiente de correlaçăo de Pearson. Modelo de regressăo: obs1 = 3.45 + 0.93 (obs3).

Gráfico 4. Regressăo para os valores de CCALHC entre os observadores 1 e 2. r: coeficiente de correlaçăo de Pearson. Modelo de regressăo: obs1 = 17.52 + 0.68 (obs2).
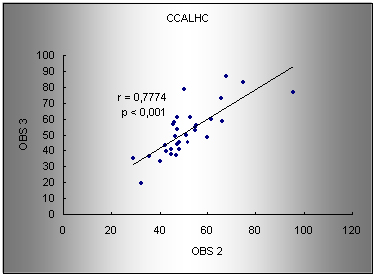
Gráfico 5. Regressăo para os valores de CCALHC entre os observadores 2 e 3. r: coeficiente de correlaçăo de Pearson. Modelo de regressăo: obs2 = 17.82 + 0.64 (obs3).

Gráfico 6. Regressăo para os valores de CCALHC entre os observadores 1 e 3. r: coeficiente de correlaçăo de Pearson. Modelo de regressăo: obs1 = 17.27+ 0.63 (obs3).
Discussăo
A maioria dos estudos de biometria hepática em crianças21,24- 26,33,34 propőe como parâmetro para avaliaçăo do tamanho do lobo hepático direito a medida do comprimento da borda anterior do fígado na linha hemi-clavicular (“liver span”), que denominamos CCALHC. Esse parâmetro, no entanto, utiliza a interface hepato-diafragmática anterior como um dos reparos anatômicos. A falta de definiçăo dessa interface, pela interposiçăo de arcos costais e/ou paręnquima pulmonar, pode comprometer a precisăo da medida e resultar em discrepâncias ao se tentar reproduzi-la.15 Em estudo realizado anteriormente40 comprovamos a reprodutibilidade de CCALHC por um mesmo observador. No presente estudo, porém, os resultados năo mostraram reprodutibilidade deste parâmetro por observadores diferentes.
O novo parâmetro que introduzimos para avaliaçăo do comprimento hepático na linha hemi-clavicular (CCPLHC), tem como referęncia a interface hepato- diafragmática posterior. Esta interface, pela ausęncia de interposiçăo de paręnquima pulmonar, mostra-se nítida, permitindo definiçăo precisa dos limites do órgăo. CCPLHC mostrou-se reprodutível, tanto em estudo de variabilidade intra- observador,40 como neste de variabilidade inter-observadores, devido ŕ definiçăo precisa de seus pontos de referęncia.
Conclusăo
Pelo apresentado, concluímos que o novo parâmetro de hepatometria em crianças é reprodutível por observadores diferentes, indicando que pode ser utilizado na determinaçăo das dimensőes do fígado em crianças e apresenta vantagens em relaçăo ao parâmetro majoritariamente utilizado em outros métodos.
Presentemente estamos realizando estudo populacional para a determinaçăo ultra- sonográfica dos valores normais do tamanho do fígado de crianças em idade pré- escolar na cidade de Săo Paulo (Brasil) aplicando o método testado.
Los autores no manifiestan
"conflictos de interés"
BIBLIOGRAFÍA
-
Walker WA, Mathis RK. Hepatomegaly. Pediatric Clin North Am, v. 22, p. 929-
42, 1975.
-
Bricks LF, Kobinger ME, Rańna W. Hepatoesplenomegalia In: Marcondes E.
Pediatria Básica 8a ed. Săo Paulo, Savier, 1991, p. 193-197.
-
Sullivan S, Krasner N, Willians R. The clinical estimation of liver size: a
comparison of thechniques and an analysis of the source of error. British Medical
Journal, v. 2, p. 1042-3, 1976.
-
Lawson EE, Grand RJ, Neff RK, Cohen LF. Clinical estimation of liver span in
infants and children. Am J Dis Child, v. 132, p. 474-6, 1978.
-
Castell DO, O'Brien KD, Munch H, Chalmers T. Estimation of liver size by
percussion in normal individuals. Ann Intern Med, v. 70, p. 1183-1189.
-
Younoszai MK, Mueller S. Clinical assesment of liver in normal children. Clin
Pediatr, v. 14, p. 378-80, 1975.
-
Weisman LE, Cagle N, Mathis R, Merenstein GB. Clinical estimation of liver size
in the normal neonate. Clin Pediatr (Phila), v. 21, p. 596-598, 1982.
-
Carpentieri U, Gustavson LP, Leach TM, Bruce H. Liver size in normal infants
and children. South Med J, v. 70, p. 1096-1097, 1977.
-
Deligeorgis D, Yannakos D, Panatotou P, Doxiadis S. The normal borders of the
liver in infancy and childhood - clinical and x-ray study. Arch Dis Child, v. 45, p.
702-704, 1970.
-
Walk L. Roengenologic determination of the liver volume. Acta Radiol, v. 55, p.
49-56, 1961.
-
Walk L. Assessment of liver size. Digestion, v. 1, p. 289-295, 1968.
-
Walk, L. Quantitative method to determine liver size. Radiologe, v. 18, p. 354-
355, 1078.
-
Walk L. Normal liver size as determined with quantitative methods. Radiologe,
v. 22, p. 188-189, 1982.
-
Walk L. Liver size in children determined with quantitative methods. Radiologe,
v. 25,p. 221-223, 1985.
-
Holder LE, Strife J, Padikal TN, Perkins PJ, Kereiakes JG. Liver size
determination in pediatrics using sonographac and scintigraphic techniques.
Radiology, v. 117, p. 349-53, 1975.
-
Markisz JA, Treves ST, Davis RT. Normal hepatic and splenic size in children:
scintigraphic determination. Pediatr Radiol, v.17, p. 273-276, 1987.
-
Naftalis J, Leevy CM. Clinical estimation of liver size. Am J Diag Dis, v. 8, p.
236-243, 1963.
-
Peternel WW, Schaefer JW, Shiff L. Clinical evaluation of liver size and hepatic
scintiscan. Am J Dis Child, v. 11, p. 346-50, 1966.
-
Rosenfield AT, Scheider PB. Rapid evaluation of hepatic size on radioisotope
scan. J Nucl Med, v.15 , p. 237-240, 1974.
-
Skrainka B, Stahlhut J, Fullbeck CL, Knight F, Holmes RA, Butt JH. Measuring
liver span: bedside examination versus ultrasound and scintiscan. J Clin
Gastroenterol, v. 8, p. 267-270, 1986.
-
Assadamongkol K, Phuapradit P, Udompanich O, Varavithya W. Liver size and
serum alkaline phosphatase in normal Thai school - aged children. J Med Assoc
Thai, v. 72, suplplement 1, p. 88-93, 1989.
-
Baddeley H, Benson M, Liefman G, Singcharoen T et al. Measurements of liver
volume using water delay ultrasonography. Diagn Imag Clin Med, v. 55, p. 330-
336, 1986.
-
Carr D, Duncan JG, Railton R, Smith CB. Liver volume determination by
ultrasound: a feasebility study. Br J Radiol, v. 49, p. 776-778, 1976.
-
Chen CM, Wang JJ. Clinical and sonographic assessment of liver size in normal
chinese neonates. Acta Paediatr, v. 82, p. 345-347, 1993.
-
Dittrich M, Milde S, Dinkel E, Baumann W, Weitzel D. Sonografic biometry of
liver and spleen in children. Pediatr Radiol, v. 13 , p. 349-353, 1983.
-
Friis H, Ndhlovu P, Mduluza T et al. Ultrasonographic organometry: liver and
spleen among children in Zimbabwe. Trop Med Int Health, v. 1, p. 183-190, 1996.
-
Gosink BB, Leymaster CE. Ultrasonic determination of hepatomegaly. J Clin
Ultrasound, v. 9, p. 37-41, 1981.
-
Haddad-Zebouni S, Hindy R, Slaba S et al. Évaluation échographique de la taille
des reins, du fois et de la rate chez l'enfant. Arch Pédiatr, v. 6, p. 1266-1270,
1999.
-
Hessel G. Hepatometria na infância - comparaçăo entre o método clínico e ultra
- sonográfico. Campinas, 1991. Dissertaçăo (Mestrado) - Faculdade de Cięncias
Médicas da Universidade Estadual de Campinas - SP.
-
Holmes JH, Sundgren C, Ilke D, Finch J. A simple ultrasonic method for
evaluating liver size. J Clin Ultrasound, v. 5, p. 89-91, 1977.
-
Jungthirapanich J, Kaewtubtim J, Poovorawan Y. A new reference line for
measuring the liver size in healthy newborns. J Assoc Thai, v. 81, p. 038-043,
1998.
-
Kardel T, Holm HH, Rasmussen SN, Mortensen T. Ultrasonic determination of
liver and spleen volumes. Scand. J Clin Lab Invest, v.27, p. 123-128, 1971.
-
Konus Ö, Özdemir A, Akkaya A, Erbas G, Celik H, Isik S. Normal liver, spleen,
and kidney dimensions in neonates, infants, and children: evaluation with
sonography. A J R, v. 171, p. 1693-1698, 1998.
-
Niederau C, Sonnenberg A, Müller JE, Erckenbrecht JF, Scholten T, Fritsch WP.
Sonographic measurements of the normal liver, spleen, pancreas, and portal vein.
Radiology, v. 149, p. 537-540, 1983.
-
Phuapradit P, Assadamongkol K, Udompanich O, Varavithya W. Liver size and
serum alkaline phosphatase in normal Thai school - aged children. J Med Assoc
Thai, v. 69, suplplement 2, p. 69-76, 1986.
-
Rasmussen, S N. Liver volume determination by ultrasonic scanning. Dan Med
Bull, v. 25, p. 1-46, 1978.
-
Rylance GW, Moreland TA, Cowan MD, Clark DC. Liver volume estimation using
ultrasound scanning. Arch Dis Child, v. 57, p. 283-286, 1982.
-
Wladimiroff JW, Sekeris A. Ultrasonic assessment of liver size in the newborn. J
Clin Ultrasound, v. 5, p. 316-320, 1977.
-
Jones JH, Robinson PJ. Organ volume determination by CT scanning: reduction
of respiration induced erros by feed back monitoring. J Comput Assist Tomogr, v.
10, p. 167-171, 1986.
-
Rocha SMS, Oliveira IRS, Widman A, Chisman BSK, Fukushima JT, Oliveira
LAN, Cerri GG. Hepatometria ultra-sonográfica em crianças (proposta de novo
método). Radiol Bras, v.36, p. 63-70, 2003.
|