El carcinoma de células pequeńas (CCP) es una neoplasia que puede aparecer en el tracto urinario, aunque ésta no es su localización más frecuente. La mayor parte asienta en la vejiga,1 si bien supone un 0.5% a un 0.7% del total de tumor es malignos de este órgano.2,3 La primera descripción en la literatura corresponde a 1975. La casuística publicada refleja una incidencia predominante en el sexo masculino (5.1:1), y una media de edad de 68 ańos.4
Histogénesis
El origen de estas neoplasias resulta controvertido, en este contexto se han formulado tres teorías principales5 acerca de su histogénesis:
a)
Derivación a partir de determinadas células neuroendocrinas, situadas cerca de la lámina basal de los endotelios, constituyentes del denominado sistema APUD6 (acrónimo anglosajón de amine precursor uptake and decarboxylation). Las células APUD, caracterizadas por poseer densos gránulos neurosecretores intracitoplasmáticos, también fueron identificadas en el carcinoma de células pequeńas vesical.7 Esta teoría, no obstante, queda cuestionada ante la evidencia de caso s en los que coexiste este tipo de neoplasia junto a un carcinoma transicional (CT).5
b)
Metaplasia surgida a partir de otras neoplasias malignas de alto grado. Esta teoría explicaría aquellos casos de coexistencia de otros tipos histopatológicos neoplásicos asociados al CCP.8
c)
Origen a partir de una célula madre pluripotencial.2 Esta teoría permitiría explicar la coincidencia de neoplasias de diferente estirpe histológica, así como el carácter heterogéneo puesto de manifiesto al analizar los patrones de ti nción inmunohistoquímica del CCP.
A pesar de la baja frecuencia descrita (14%) de carcinoma in situ,1 se ha sugerido para el CCP la posibilidad de un origen urotelial, si se toma como base el alto porcentaje de expresión de citoqueratina: positividad de CAM 5.2 en un 64 % de los casos.6
Genética
Estudios de hibridación comparativa reflejan la existencia de un gran número de cambios citogenéticos en el CCP vesical.9 Las modificaciones descritas con mayor frecuencia incluyen deleciones a nivel cromosómico 10q, 4q, 5q y 13q; así como adi ciones en las localizaciones 8q, 5p, 6p y 20q. Deleciones en los cromosomas 4, 5q, 6q, 11p y 13q, así como adiciones en 17q fueron verificadas tanto en el CCP como en el CT vesical, y además, en los casos de coexistencia de ambos tipos de neoplasia, toda s las alteraciones cromosómicas presentes en el CT se encontraron asimismo en el CCP, lo cual supondría un apoyo para la teoría ya citada de la histogénesis a partir de una célula pluripotencial. Estudios10 en tumores mixtos acerca de la pérdi da de heterozigosidad –e inactivación del cromosoma X en pacientes mujeres– ofrecen asimismo resultados en dicha línea.
La existencia de áreas de amplificación genómica, sugestivas de actividad oncogénica, fue descrita en las localizaciones 1p22-32, 3q26.3, 8q24 (incluye el oncogén CMYC) y 12q14-21 (incluye el oncogén MDM2).9 Otros estudios citogenéticos demost raron asimismo alteraciones complejas en los cromosomas 9, 11 y 18, así como la sobreexpresión –en un 77% de los casos11– del gen p53.
Diagnóstico clínico y exploraciones complementarias
Las manifestaciones clínicas del CCP son de carácter inespecífico, ejemplo de lo cual es la hematuria monosintomática, el signo de debut más frecuentemente referido en la literatura (90% de los casos).12 Síntomas de presentación asimismo f recuente son disuria, polaquiuria y molestias hipogástricas y/o pelvianas.13
Los estudios por imágenes –urografía intravenosa y ecografía, entre otras– no permiten el diagnóstico diferencial con otras neoformaciones, como por ejemplo el CT. Las imágenes cistoscópicas tampoco aportan características específicas, aunque ponen de manifiesto la existencia de grandes lesiones –el diámetro medio en algunas series2 fue de 5.5 cm–, de carácter más o menos sólido, nodular, generalmente aisladas, con posible componente ulcerativo-necrótico y áreas hemorrágicas superficiales, as í como capacidad infiltrativa parietal. Las localizaciones topográficas vesicales más frecuentes del CCP2,5 son las paredes laterales (54%) y posterior (20% de los pacientes), trígono (10%), cúpula (8%) y pared anterior (8%). El hallazgo de es te tipo de neoplasia en el interior de un divertículo fue descrita hasta en 4.7% de los casos.14
Diagnóstico anatomopatológico
El estudio histopatológico de los fragmentos de resección transuretral, así como de la pieza de cirugía radical –en su caso– constituye la base necesaria sobre la que se sustenta la filiación del CCP vesical (figura 1). Existen tres tipos de CCP, de acuerdo con las características apreciadas al microscopio óptico:5,8,15
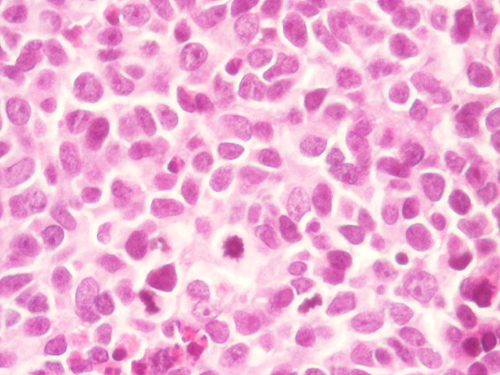
Figura 1.
Preparación histopatológica correspondiente a carcinoma vesical de células pequeńas (hematoxilina-eosina, x 100).
a)
El tipo oat cell o de células en “copo de avena”, constituido por pequeńas células redondeadas, de núcleo picnótico circular u oval, con nucléolo poco evidente y escaso citoplasma:16 hallazgos confirmados también mediante microscopio electrónico.17,18
b)
El tipo de células “intermedias”, en el que las células son de mayor tamańo, fusiformes o poligonales, y con un mayor componente citoplasmático.
c)
En hasta el 38% al 50% de los casos4,19 se aprecia el patrón “celular combinado”, en el que otra neoplasia diferente se asocia al CCP, mezclándose ambas entre sí12 de forma difusa o focal; el CT es la más frecuente; con mucha menor incidencia de adenocarcinomas y carcinomas escamosos y, excepcionalmente, tumor carcinoide y sarcoma.5 La variedad histológica predominante del CCP en estos pacientes es el de células “intermedias”.
La descripción arquitectural más frecuente del CCP vesical6,20 la constituyen láminas difusas o “moldeados” celulares con formaciones aisladas trabeculares o en cinta; es frecuente la descripción de mitosis e invasión vascular, así como –en la mitad de los casos–necrosis individualizada, que origina un aspecto de “cielo estrellado”. La fragilidad celular es causa de imágenes con artefactos (squash), en las que se aprecian pequeńas formaciones en banda, de color azul. Asimismo está descrito el denominado “fenómeno de Azzopardi”, consistente en la aparición de depósitos tisulares perivasculares de material basófilo (DNA).
Los TC se diferencian de los CCP por una disposición en agrupaciones o nidos celulares, con mayor pleomorfismo y presencia de nucléolos prominentes.21 Otras neoplasias que entran a formar parte del diagnóstico diferencial, como el linfoma, las constituyen células hipercromáticas redondeadas, como en el CCP, si bien las mitosis resultan infrecuentes y el componente necrótico está ausente.16
La microscopia electrónica también ha sido utilizada en casos de dificultad para la diferenciación del CCP con respecto a otras variedades.2,17,18 El hallazgo más relevante consiste en la presencia de gránulos neurosecretores intracitoplasmáti cos –centrales o periféricos– de núcleo denso y de 30 a 300 nm de diámetro. El CCP se diferencia asimismo por la ausencia de tonofilamentos –al contrario que el carcinoma escamoso–, gránulos intracitoplasmáticos de mucina y microvilli –al contrario que el adenocarcinoma–.5
Inmunohistoquímica
Existe una amplia variedad de marcadores expresados por el CCP que pueden clasificarse en epiteliales y neuroendocrinos. De los primeros, los más frecuentes4 son el antígeno carcinoembrionario (CEA) –en el 57% de los pacientes– y el antígeno e pitelial de membrana (EMA) (56%), si bien ninguno de ellos tiene carácter específico.1 La citoqueratina (CAM 5.2) se verifica en 25% de los casos,22 y su característico patrón de tinción (punteado perinuclear) permite una gran discriminación con respecto al CT, puesto que en éste último el patrón es membranoso.23
La mayor parte de los CCP expresan al menos dos marcadores neuroendocrinos diferentes1,5 (figura 2).
El más frecuente es la enolasa neuronal específica (NSE), positiva en aproximadamente 90% de los casos,2,5-8,12,15,20 si bien se encuentra asimismo presente en el 76% de los CT de alto grado.5 La sinaptofisina y cromogranina A resultan positivas en 30% a 50% de los pacientes,1 lo que demuestra un alto grado de especificidad para ambos con respecto al CCP .

Figura 2.
Positividad de marcadores neurohistoquímicos: enolasa neuronal específica (x 200) (derecha) y sinaptofisina (x 200) (izquierda).
Los estudios comparativos22,24 acerca de la expresión de la glicoproteína transmembrana CD44v6 en el CCP y en el CT vesical dieron como resultado una diferencia en la incidencia de aparición del 7% frente al 60% a favor del segundo, por lo que constituye una nueva herramienta de potencial utilidad para la diferenciación de ambos tipos de neoplasia.
En el diagnóstico diferencial del CCP primario vesical deben considerarse otros procesos neoplásicos. Entre ellos se encuentran el infrecuente CCP secundario,25 los carcinomas plasmocitoides, los linfoepitelioides, y los linfomas –ya citados–.
En este sentido, el uso de tinciones específicas para el antígeno común leucocitario (LCA) resulta de utilidad, dado que es característico de los procesos linfoproliferativos, sin que haya sido descrito hasta el momento en el CCP.12
Historia natural
Hasta en 94% de los casos el CCP vesical presenta invasión muscular en el momento del diagnóstico.6 La enfermedad metastásica fue descrita en un 56% a 67% de los pacientes con dicha neoplasia, sus localizaciones más frecuentes son:1,3,5,1 6,24 ganglios linfáticos (56%), hueso (44%), hígado (33%) y pulmón (20%).
Un signo de enfermedad metastásica es la neuropatía periférica sensorial, que es considerada una manifestación paraneoplásica como consecuencia de la producción de autoanticuerpos antineuronales; en este sentido, la presencia de lgG anti-HU demostró ser específico del marcador.26 Otros hallazgos5,27,28 implicados en el síndrome paraneoplásico asociado al CCP vesical son alteraciones electrolíticas como hipercalcemia o hipofosfatemia, y la secreción ectópica de ACTH.
Factores pronósticos
El CCP vesical es una neoplasia clínicamente agresiva, dada su predisposición a la infiltración parietal y a afectar la vasculatura. Algunos autores seńalan cifras de supervivencia media del 8% a los cinco ańos.1,5
El pronóstico de este tipo de tumores se vincula clásicamente al estadio clínico presente;1,14 así, la revisión de la casuística de la Clínica Mayo29 seńala supervivencias a cinco ańos, para pacientes con neoplasia en estadios II, I II y IV, de 63.6%, 15.4% y 10.5%, repectivamente. Sin embargo, en algunas publicaciones3,30 se sugirió que este factor no sería independiente, en tanto existe la probabilidad de micrometástasis ya presentes en el momento del diagnóstico, incluso en pacientes con enfermedad aparentemente confinada. En el único estudio prospectivo publicado hasta la fecha,31 las diferencias en cifras de supervivencia entre enfermedad limitada y diseminada –definida la primera como cualquier estadio local con afección locorregional de un solo ganglio linfático como máximo, menor de 2 cm de diámetro–, no resultaron significativas, si bien el poder estadístico del análisis es limitado dado su pequeńo tamańo muestral.
Otros factores predictivos de mal pronóstico serían la enfermedad metastásica confirmada ya en el momento del diagnóstico inicial, y la edad del paciente superior a 65 ańos.
No se encontraron diferencias estadísticamente significativas en cuanto a cifras de supervivencia al comparar las distintas variedades histológicas del CCP.8 Se desconoce actualmente el valor predictivo de los cambios citogenéticos mendionados con anterioridad en este artículo. En este sentido, la detección del p53 no pudo ser establecida como un factor de peor pronóstico.1
Tratamiento
Cirugía (con quimioterapia o radioterapia complementaria o sin ellas)
Las opciones de tratamiento quirúrgico permiten diferentes plateamientos intervencionistas según los autores; así, existen enfoques terapéuticos basados en la cistectomía, radical12,19 o parcial,32 o en la resección transuretral radical.1 El enfoque quirúrgico aislado ha proporcionado, sin embargo, resultados insatisfactorios. Así quedó reflejado en series como la de Trías,1 en la que la supervivencia de los pacientes sometidos únicamente a cistoprostatectomía radical (estadios II-III) osciló entre 1 y 10 meses. Otros autores5,33 coinciden también en seńalar una alta tasa de recurrencias si no se adoptan estrategias terapéuticas complementarias a la cirugía. Alguna publicación,34 no obstante, aporta supervivencias discretamente más prolongadas, con un rango de 11 a 25 meses. Aun así, algunos autores,29 sobre la base de estudios retrospectivos, creen que en el estadio II puede prescindirse de la quimioterapia adyuvante.
La tendencia más generalizada está definida por la adopción de estrategias combinadas con la cirugía basadas en la quimioterapia complementaria, en la que el cisplatino se perfila como el agente antineoplásico que permite un incremento significativo en l a supervivencia de estos pacientes, hecho confirmado por algunas publicaciones30 mediante análisis multivariado. En un estudio prospectivo,31 los análisis de regresión atribuyeron al uso de quimioterapia un valor como factor pronóstico independiente positivo con vistas a la supervivencia.
Abbas5 observó en pacientes tratados con quimioterapia adyuvante una supervivencia del 73% tras un seguimiento medio de 21.1 meses. En una serie4 de 18 pacientes sometidos a la misma orientación terapéutica se registró asimismo una supervivencia del 73%, con una media temporal de 27 meses. En una serie con menor casuística12 se trató a los sujetos con CCP puro con una combinación adyuvante de adriamicina, etopósido y cisplatino, mientras que a los que presentaron coexist encia de CCP y CT se los sometió a tratamiento con MVAC –asimismo adyuvante–; con lo que se obtuvo en conjunto una supervivencia del 100% tras 34 meses de seguimiento medio. Oesterling19 también aporta una supervivencia del 100% en pacientes con tumores mixtos tratados con MVAC adyuvante tras un seguimiento medio de 21 meses. Nuestra experiencia35 con quimioterapia adyuvante, basada en la combinación de carboplatino más etopósido resultó satisfactoria, con una supervivencia libre d e enfermedad de hasta 48 meses.
Algunos artículos reflejan resultados satisfactorios basados en la administración de quimioterapia neoadyuvante. Así, Cheng36 presenta el ejemplo de un paciente con metástasis en cadenas linfáticas pelvianas que fue tratado con una combinación de metotrexato + vincristina + adriamicina + ciclofosfamida (MVAC), y sometido posteriormente a cistoprostatectomía radical, libre de enfermedad a los 9 ańos de seguimiento. La literatura37 refleja el caso de un paciente con CCP músculo-invasivo que fue asimismo tratado con MVAC neoadyuvante seguido de cistoprostatectomía radical, sin evidencia de recidiva de la enfermedad a los 3 ańos poscirugía. Walther38 seńala que de sus 7 pacientes tratados con quimioterapia –en 5, de forma neoadyuvante– más cistectomía, 5 están libres de enfermedad tras un seguimiento de 36 meses. La casuística del M. D. Anderson Cancer Center33 refleja cifras de supervivencia específicas de cáncer del 78% con quimioterapia previa a la cistoprostatectomía radical, tras cinco ańos de seguimiento, haciendo especial hincapié en la necesidad de realizar estudios prospectivos con suficiente número de pacientes para establecer las mejores directrices de tratamiento en el futuro.
La cistectomía parcial, complementada por quimioterapia, radioterapia o ambas, también fue utilizada como estrategia terapéutica en el CCP. Ejemplo de ello es una serie39 –aunque con número de pacientes muy limitado– de tumores en estadio III, en la que se obtuvo una supervivencia libre de enfermedad de hasta 78 meses tras la irradiación adyuvante.
Los resultados luego de la resección transuretral (RTU) aislada son generalmente escasos. La literatura seńala cifras medias de supervivencia de entre 3 y 7 meses.1,40
Preservación vesical. Radiación sola o con quimioterapia complementaria
La supervivencia media de los casos publicados de CCP vesical tratados únicamente con radioterapia tras biopsia endoscópica diagnóstica no sobrepasa los ocho meses. La combinación de quimioterapia y radioterapia parece ofrecer mejores resultados. En una serie41 se administró un protocolo de seis ciclos de etopósido + cisplatino, alternados con la combinación de ciclofosfamida + doxorrubicina + vincristina. Cuatro de los 5 pacientes presentaron respuesta completa confirmada por biopsia cistoscópica, los cuales fueron sometidos posteriormente a irradiación externa (45 Gy en pelvis, 60 Gy en vejiga), y se obtuvo una supervivencia del 100% tras un seguimiento medio de 44 meses, si bien en uno de los casos (tumor primario de patrón “celular combinado”) se recurrió a cistoprostatectomía radical de rescate tras confirmarse recidiva local –por CT– a los 12 meses de seguimiento posradioterapia. Lohrisch13 informó, para una serie de 10 pacientes con enfermedad localizada, 70% y 44% de supervivencia a 2 y 5 ańos, respectivamente, tras el tratamiento integrado con quimioterapia (etopósido + cisplatino) y radioterapia local externa. Bex31 intenta establecer un paralelismo entre los regímenes terapéuticos de los CCP pulmonar y vesical, e informa 64.7% de respuestas completas para aquellos casos con neoplasia urológica limitada, sometidos a cuatro ciclos de la combinación etopósido + cisplatino, más radioterapia secuencial (60 Gy de dosis media). A este respecto, cabe seńalar que u n nuevo agente quimioterapéutico, el irinotecán (inhibidor de la enzima topoisomerasa tipo I), se perfila como una opción de combinación con cisplatino (en sustitución del etopósido) para el CCP pulmonar, dados los mejores resultados obtenidos en estudio s comparativos42 (fase II) con respecto al porcentaje medio de supervivencia a dos ańos, así como a la menor incidencia de mielosupresión y diarrea graves. Por lo tanto podría en el futuro ser parte de los regímenes utilizados también en el CC P vesical. Otras comunicaciones con casos puntuales43 informan asimismo supervivencias libres de enfermedad de hasta 4.5 ańos en CCP en estadio III, tras la combinación de quimioterapia (cisplatino + metotrexato + vinblastina) seguida de irradiación externa.
La radioterapia profiláctica craneal no está indicada en el CCP,31,33 dado que a pesar de estar descrita en la literatura, la incidencia de enfermedad metastásica cerebral es muy reducida.
Coincidimos con otros autores44 en la necesidad de realizar futuros estudios multicéntricos, cuyo carácter prospectivo y aleatorizado permita mayor discriminación positiva de alguna estrategia terapéutica por sobre el resto, dentro del ámbito de la medicina basada en la evidencia.
BIBLIOGRAFÍA
1. Trías I, Algaba F, Condom E, y col.
Small cell carcinoma of the urinary
bladder. Presentation of 23 cases and review of 134 published cases.
Eur Urol 2001; 39: 85-90.
2. Blomjous CEM, Vos W, De Voogt HJ, Van der Valk P, Meijer CJLM.
Small cell carcinoma of the urinary bladder. A clinicopathologic,
morphologic, immunohistochemical and ultrastructural study of 18
cases. Cancer 1989; 64:1347-57.
3. Holmang S, Borghede G, Johansson SL. Primary small cell carcinoma
of the bladder: a report of 25 cases. J Urol 1995; 153:1820-2.
4. Sved P, Gómez P, Manoharan M, Civantos F, Soloway MS. Small cell
carcinoma of the bladder. B J U Int 2004; 94:12-7.
5. Abbas F, Civantos F, Benedetto P, Soloway MS. Small cell
carcinoma of the bladder and prostate. Urology 1995; 46:617-30.
6. Ali SZ, Reuter VE, Zakowski MF. Small cell neuroendocrine
carcinoma of the urinary bladder. A clinicopathologic study with
emphasis on cytologic features. Cancer 1997; 79:356-61.
7. Pearse A. The cytochemistry and ultrastructure of polypeptide
hormone-producing cells of the APUD series and the embryologic,
physiologic, and pathologic implications of the concepts. J
Histochem Cytochem 1969; 17:303-13.
8. Christopher ME, Seftel AD, Sorenson K, Resnick M. Small cell
carcinoma of the genitourinary tract: an immunohistochemical,
electron microscopic and clinicopathological study. J Urol 1991;
146:382-8.
9. Terracciano L, Richter J, Tornillo L, y col. Chromosomal
imbalances in small cell carcinomas of the urinary bladder. J Pathol
1999; 189:230-5.
10. Cheng L, Jones TD, McCarthy RP, y col. Molecular genetic
evidence for a common clonal origin of urinary bladder small cell
carcinoma and coexisting urothelial carcinoma. Am J Pathol 2005;
166:1533-9.
11. Atkin NB, Baker MC, Wilson GD. Chromosome abnormalities and p53
expression in a small cell carcinoma of the bladder. Cancer Genet
Cytogenet 1995; 79:111-4.
12. Grignon DJ, Ro JY, Ayala AG, y col. Small cell carcinoma of the
urinary bladder. A clinicopathologic analysis of 22 cases. Cancer
1992; 69:527-36.
13. Lohrisch C, Murray N, Pickles T. Sullivan L. Small cell
carcinoma of the bladder: long term outcome with integrated
chemoradiation. Cancer 1999; 86:2346-52.
14. Angulo JC, López JI, Sánchez Chapado M, y col. Small cell
carcinoma of the urinary bladdder. A report of two cases with
complete remission and a comprehensive literature review with
emphasis on therapeutic decisions. J Urol Pathol 1996; 5:1-19.
15. The World Health Organization histological typing of lung
tumours. Second edition. Am J Clin Pathol. 1982; 77:123-36.
16. Acs G, Gupta PK, Baloch ZW. Cytomorphology of high-grade
neuroendocrine carcinoma of the urinary tract. Diagn Cytopathol
2000; 23:92-6.
17. Kim CK, Lin JI, Tseng CH. Small cell carcinoma of urinary
bladder. An ultrastructural study. Urology 1984; 24:384-6.
18. Ordóńez NG, Khorsand J, Ayala AG, Sneige N. Oat cell carcinoma
of the urinary tract. An immunohistochemical and electron
microscopic study. Cancer 1986; 58:2519-30.
19. Oesterling JE, Brendler CB, Burgers JK, Marshall FF, Epstein JI.
Advanced small cell carcinoma of the bladder. Successful treatment
with combined radical cystoprostatectomy and adjuvant methotrexate,
vinblastine, doxorubicin, and cysplatin chemothera
py. Cancer
1990; 65:1928-36.
20. Mills SE, Wolf JT 3rd, Weiss MA, y col. Small cell
undifferentiated carcinoma of the urinary bladder. A light
microscopic, immunohistochemical and ultrastructural study of 12
cases. Am J Surg Pathol 1987; 11:606-17.
21. Shin HJC, Caraway NP. Fine-needle aspiration biopsy of
metastatic small cell carcinoma from extrapulmonary sites. Diagn
Cytopathol 1998; 19:177-81.
22. Iczkowski KA, Shanks JH, Allsbrook WC, y col. Small cell
carcinoma of urinary bladder is differentiated from urothelial
carcinoma by chromogranin expression, absence of CD44 variant 6
expression, a unique pattern of cytokeratin expression, and more i
ntense g -enolase
expression. Histopathology 1999; 35:150-6.
23. Cheng C, Nicholson A, Lowe DG, Kirby RS. Oat cell carcinoma of
urinary bladder. Urology 1992; 39:504-7.
24. Iczkowski
KA, Shanks JH, Bostwick DG. Loss of CD44 variant 6 expression
differentiates small cell carcinoma of urinary bladder from
urothelial (transitional cell) carcinoma. Histopathology 1998;
32:322-7.
25. Di Pietro M, Zeman RK, Kehoane M, Rosenfield AT. Oat cell
carcinoma metastatic to ureter. Urology 1983; 22:419-20.
26. Anderson NE, Rosemblum MK, Graus F, Wiley RG, Posner JB.
Autoantibodies in paraneoplastic syndromes associated with
small-cell lung cancer. Neurology 1988; 38:1391-8.
27. Partanen S, Asikainen U. Oat cell carcinoma of the urinary
bladder with ectopic adrenocorticotropic hormone production. Hum
Pathol 1985; 16:313-5.
28. Reyes CV, Soneru I. Small cell carcinoma with hypercalcemia.
Cancer 1985; 56:2530-33.
29. Choong NW, Quevedo JF, Kaur JS. Small cell carcinoma of the
urinary bladder. The Mayo Clinic experience. Cancer 2005;
103:1172-8.
30. Mackey JR, Au HJ, Hugh J, Venner P. Genitourinary small cell
carcinoma: determination of clinical and therapeutic factors
associated with survival. J Urol 1998; 159:1624-9.
31. Bex A, Nieuwenhuijzen JA, Kerst M, y col. Small cell carcinoma
of bladder: a single-center prospective study of 25 cases treated in
analogy to small cell lung cancer. Urology 2005; 65:295-9.
32. Ripa Saldías L, Delpón Pérez E, Fernández Rosáenz J, Córdoba
Iturriagagoitia A, Monzón Muńoz FJ, Ruiz de Azúa Ciria Y. Carcinoma
de células pequeńas de vejiga. Aportación de un nuevo caso. Actas
Urol Esp 1997; 21:495-500.
33. Siefker-Radtke AO, Dinney CP, Abrahams NA, y col. Evidence
supporting preoperative chemotherapy for small cell carcinoma of the
bladder: a retrospective review of the M. D. Anderson cancer
experience. J Urol 2004; 172:481-4.
34. López JI, Angulo JC, Flores N, Toledo JD. Small cell carcinoma
of the urinary bladder. A clinicopathological study of six cases. Br
J Urol 1994; 73:43-9.
35. Cruz Guerra NA, Fradejas Rodríguez A, Zamora Martínez T, del
Valle Manteca A, Tinajas Saldańa A, Tarroc Blanco A. Carcinoma
vesical infiltrante de células pequeńas. Descripción de un nuevo
caso tratado con cirugía radical más quimioterapia. Arch Esp Urol
2004; 57:854-6.
36. Cheng D, Unger P, Forscher CA, Fine E. Successful treatment of
metastatic small cell carcinoma of the bladder with methotrexate,
vinblastine, doxorubicin and cisplatin therapy. J Urol 1995;
153:417-9.
37. Nejat RJ, Purohit R, Goluboff ET, Petrylak D, Rubin MA, Benson
MC. Cure of undifferentiated small cell carcinoma of the urinary
bladder with M-VAC chemotherapy. Urol Oncol 2001; 6:53-5.
38. Walther PJ. Adjuvant/ neoadjuvant etoposide/ cisplatin and
cystectomy for management of invasive small cell carcinoma of the
bladder. J Urol 2002; 167 Suppl:285.
39. Podesta AH, True LD. Small cell carcinoma of the bladder: report
of five cases with immunohistochemistry and review of the literature
with evaluation of prognosis according to stage. Cancer 1989;
64:710-4.
40. Helpap B. Morphology and therapeutic strategies for
neuroendocrine tumours of the genitourinary tract. Cancer 2002;
95:1415-20.
41. Bastús R, Caballero JM, González G, y col. Small cell carcinoma
of the urinary bladder treated with chemotherapy and radiotherapy:
results in five cases. Eur Urol 1999; 35:323-6.
42. Noda K, Nishiwaki Y, Kawahara M, y col. Irinotecan plus
cisplatin compared with etoposide plus cisplatin for extensive
small-cell lung cancer. N Engl J Med 2002; 346:85-91.
43. Oblon DJ, Parsons JT, Zander DS, Wajsman Z. Bladder preservation
and durable complete remission of small cell carcinoma of the
bladder with systemic chemotherapy and adjuvant radiation therapy.
Cancer 1993; 71:2581-4.
44. Muńoz Vélez D, García-Miralles Grávalos R, Amengual Antich I,
Benejam Gual JM. Carcinoma vesical de células pequeńas. Aportación
de un nuevo caso y revisión de la literatura. Actas Urol Esp 2002;
26:811-5.
|



